")
")
| Issue |
Med Sci (Paris)
Volume 26, Number 6-7, Juin–Juillet 2010
|
|
|---|---|---|
| Page(s) | 561 - 563 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/2010266-7561 | |
| Published online | 15 juin 2010 | |
Le facteur d’échange Arhgef1 : une nouvelle cible dans l’hypertension artérielle
The exchange factor Arhgef1: a new target in hypertension
UMR-S915, IRT-UN, Institut du thorax, 8 quai Moncousu, 44007 Nantes, France
* Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
* Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Les maladies cardiovasculaires, avec en tête l’infarctus du myocarde, l’in-suffisance cardiaque et les accidents vasculaires cérébraux, représentent en France, comme dans bien d’autres pays développés, les principales causes de mortalité. L’hypertension artérielle (HTA) en est le facteur de risque le plus important et le plus fréquent : elle atteint 31 % de la population française adulte. Malgré des avancées thérapeutiques importantes au cours des dix dernières années, l’HTA reste aujourd’hui insuffisamment détectée, traitée et contrôlée, et les mécanismes moléculaires impliqués dans sa pathogenèse mal connus.
Tonus vasculaire et contractilité artérielle contrôlent la pression artérielle
Une des caractéristiques communes a la plupart des hypertensions est une augmentation des résistances artérielles périphériques. La contraction des cellules musculaires lisses de la paroi vasculaire (CMLV) régule le diamètre artériel et le tonus vasculaire et se présente ainsi comme un déterminant majeur des résistances périphériques. L’état contractile des CMLV reflète à chaque instant l’équilibre entre l’activité des facteurs vasoconstricteurs et vasodilatateurs qui contrôlent le niveau de phosphorylation des chaînes légères régulatrices de la myosine de type II (MLC20) [1]. Une phosphorylation accrue de la MLC20 produit une vaso-constriction et une augmentation de la pression artérielle. À l’opposé, une diminution de la phosphorylation de la MLC20 induit une vaso-dilatation et une baisse de la pression artérielle. La phosphorylation et la déphosphorylation de la MLC20 sont catalysées respectivement par deux enzymes, la kinase MLCK (myosin light chain kinase) et la phosphatase MLCP (myosin light chain phosphatase).
Bien que l’implication du Ca2+, via la stimulation de l’activité de la MLCK, soit connue depuis longtemps, la décou-verte du rôle essentiel de la protéine G monomérique RhoA dans le mécanisme de sensibilisation au Ca2+ induite par les vasoconstricteurs a constitué un progrès majeur dans la compréhension des mécanismes moléculaires de la contraction des CMLV [2, 3]. RhoA appartient à la famille des protéines Rho, elle-même membre de la super-famille Ras. C’est un commutateur moléculaire qui alterne entre un état inactif lié au GDP et un état actif lié au GTP. Sous l’action d’un stimulus, un facteur d’échange GEF (guanine nucleotide exchange factor) catalyse la substitution du GDP par le GTP qui active RhoA et lui permet d’activer à son tour ses effecteurs. De nombreux vasoconstricteurs agissant via des récepteurs à sept domaines trans-membranaires couplés aux protéines G trimériques activent RhoA selon cette cascade de réactions; c’est le cas de l’angiotensine II (Ang II), l’endothéline 1, la noradrénaline et le throm-boxane A2. La sérine-thréonine kinase Rho kinase est l’effecteur de RhoA responsable de la sensibilisation au Ca2+ lors de la contraction des CMLV [4]. Rho-kinase phosphoryle la sous-unité régulatrice MYPT (myosin phosphatase target subunit) de la MLCP etinhibe ainsi son activité (Figure 1). Ce mécanisme est indispensable à l’ef-fet contractant des vasoconstricteurs cités précédemment.
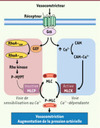 |
Figure 1 Signalisation de la contraction dans les CMLV. L’augmentation de la phosphorylation des chaînes légères de myosine (P-MLC) induit le développement de la contraction. La stimulation d’un récepteur à sept domaines transmembranaires par un vasoconstricteur produit, via les sous-unités à des protéines G trimériques (Gα) auxquelles ils sont couplés, (1) la stimulation de la kinase des MLC (MLCK) consécutive à l’augmentation du Ca2+ intracellulaire et à sa liaison à la calmoduline (CAM), et (2) l’inhibition de la phosphatase des MLC (MLCP) via la phosphorylation de la MYPT par Rho kinase. GEF : facteurs d’échange de RhoA. |
L’activation de la protéine G monomérique RhoA dans la paroi artérielle : une cause commune d’HTA
Une activation excessive de RhoA est retrouvée dans les modèles expérimentaux d’hypertension [3, 5] et chez l’homme [6]. Bien que cette hyperactivité de RhoA soit considérée comme un déterminant critique de la pathogenèse de l’HTA, son origine est inconnue. Néanmoins, puisque l’activation de RhoA par un vasocons-tricteur est dépendante de l’activation d’un GEF (Figure 1), un intérêt particulier a été naturellement porté aux GEF de RhoA exprimés dans les CMLV et à leur rôle potentiel dans l’hypertension. Les séquences ADN correspondant àenviron 60 GEF des protéines Rho ont été identifiées dans les génomes de mammifères, et 25 à 30 d’entre eux sont capables d’activer RhoA [7]. Nous avons donc émis l’hypothèse que l’hyperactivation de RhoA est un processus commun à la pathogenèse de différentes formes d’HTA, mais que les GEF responsables de cette hyper-activité pourraient être différents selon les formes d’HTA et spécifiques.
Le GEF de RhoA Arhgef1 est responsable de l’activation de RhoA par l’Ang II
Nous avons récemment examiné cette hypothèse en nous intéressant à l’hypertension dépendante de l’Ang II. L’Ang II est le principal effecteur du système hormonal rénine-angiotensine-aldostérone (SRAA) qui joue un rôleclé dans la régulation de la pression artérielle. Parmi ses nombreux effets, l’Ang II est un puissant vasoconstricteur qui contrôle directement le tonus vasculaire par l’activation des récepteurs de type 1 (AT1) exprimés sur les CMLV. L’activation du système SRAA ainsi que des taux élevés d’Ang II induisent une HTA. Nous avons donc voulu identifier le(s) GEF responsable(s) de l’activation de RhoA par l’Ang II dans les CMLV. Notre première stratégie a consisté à inhiber un à un par des ARN interférents tous les GEF de RhoA exprimés dans les CMLV. Seule l’inhibition de l’expression du GEF Arhgef1 empêchait l’activation de RhoA par l’Ang II dans les CMLV, sans modifier son activation par d’autres vaso-constricteurs comme l’endothéline-1, la phényléphrine ou le thromboxane-A2 [8]. De plus, l’inhibition de l’expression d’Arhgef1 dans des anneaux d’artères abolit la contraction induite ex vivo par l’Ang II sans affecter la contraction produite par d’autres vasoconstricteurs [8]. Ces données suggéraient donc qu’Arhgef1 est le GEF qui couple spécifiquement les récepteurs AT1 de l’Ang II à l’activation de RhoA, et que cette voie de signalisation joue un rôle essentiel dans la contraction artérielle produite par l’Ang II.
Pour valider ces résultats in vivo, nous avons développé un modèle de souris transgénique n’exprimant pas la protéine Arhgef1 spécifiquement dans les CML. L’infusion continue d’Ang II à ces souris ne modifie pas leur pression artérielle alors qu’elle provoque une HTA chez les souris contrôles (Figure 2A) [8]. En revanche, le contrôle de la pression artérielle par d’autres médiateurs reste normal chez les souris n’exprimant pas Arhgef 1 dans les CML. Ainsi, l’absence d’Arhgefl dans les CML protège de l’HTA induite par l’Ang II mais n’altère pas les autres mécanismes de régulation de la pression artérielle. Par des méthodologies similaires, l’équipe de Offermanns a montré que les souris n’exprimant pas Arhgef 12, un autre GEF de RhoA, sont résistantes à l’HTA induite par un régime riche en sel [9]. Ces travaux confirment ainsi notre hypothèse : les GEF responsables de l’hyperactivité de RhoA et de l’augmentation de la vasoconstriction varient selon l’origine de l’HTA : Arhgef1 est impliquée dans l’HTA liée à l’Ang II et Arhgef12 dans celle provoquée par un excès de sel (Figure 2B).
 |
Figure 2 Rôle essentiel de Arghef1 dans l’HTA dépendante de l’Ang II. A. Effet de l’infusion d’Ang II sur la pression artérielle systolique de souris contrôles (CML-Arhgef1lox/lox) et de souris n’exprimant pas Arhgef1 dans les CML (CMLArhgef1-KO). B. Représentation schématique du rôle respectif de Arhgef12 et Arhgef1 dans l’HTA induite par un excès de sel et l’Ang II (ETAR : récepteur A de l’endothéline; AT1R : récepteur de l’Ang II de type 1). |
Conclusion
Les résultats exposés ci-dessus apportent des éléments nouveaux dans la compréhension des mécanismes moléculaires de la régulation du tonus et de la pression artériels. La position stratégique des GEF de RhoA les identifie aujourd’hui comme des cibles thérapeutiques potentielles pour inhiber sélectivement l’hyperactivité de RhoA associée à différentes formes d’HTA. Arghef1 pourrait être une cible thérapeutique dans les HTA dépendantes de l’Ang II alors que des inhibiteurs d’Arhgef 12 pourraient être utilisés dans les HTA liées à un excès de sel. Les inhibiteurs ciblant les GEF de RhoA spécifiquement responsables de l’hyperactivité de RhoA ont l’avantage de laisser la protéine RhoA disponible. Elle peut donc être recrutée par d’autres récepteurs et d’autres voies de signalisation auxquelles ces GEF ne participent pas, préservant ainsi le contrôle physiologique de la pression artérielle par les médiateurs vasoactifs. L’avantage thérapeutique d’un inhibiteur d’Arhgef1 par rapport aux inhibiteurs des récepteurs AT1 de l’Ang II déjà largement utilisés dans le traitement de l’HTA devra cependant être démontré. Une piste à explorer est le rôle potentiel d’Arhgef1 dans les effets synergiques de l’Ang II et de l’aldostérone sur l’activité de RhoA [10]. Si Arhgef1 est impliqué dans cette synergie, les inhibiteurs d’Arhgef1 auraient alors un intérêt supplémentaire certain.
Conflit d’intérêts
Les auteurs déclarent n’avoir aucun conflit d’intérêts concernant les données publiées dans cet article.
Remerciements
Ce travail a bénéficié du soutien de l’Agence nationale de la recherche (ANR-05-PC0D-015-01 et ANR-08-GENO-040-01), et de la Fondation pour la recherche médicale (DEQ20051205767 et DEQ20090515416).
Références
- Somlyo AP, Somlyo AV. Signal transduction by Gproteins, rho-kinase and protein phosphatase to smooth muscle and non-muscle myosin II. J Physiol 2000 ; 522 : 177-85. [Google Scholar]
- Gong MC, Iizuka K, Nixon G, et al. Role of guanine nucleotide-binding proteins–ras-family or trimeric proteins or both–in Ca2+ sensitization of smooth muscle. Proc Natl Acad Sci USA 1996 ; 93 : 1340-5. [Google Scholar]
- Uehata M, Ishizaki T, Satoh H, et al. Calcium sensitization of smooth muscle mediated by a Rhoassociated protein kinase in hypertension. Nature 1997 ; 389 : 990-4. [Google Scholar]
- Loirand G, Guerin P, Pacaud P. Rho kinases in cardiovascular physiology and pathophysiology. Circ Res 2006 ; 98 : 322-34. [Google Scholar]
- Seko T, Ito M, Kureishi Y, et al. Activation of RhoA and inhibition of myosin phosphatase as important components in hypertension in vascular smooth muscle. Circ Res 2003 ; 92 : 411-8. [Google Scholar]
- Masumoto A, Hirooka Y, Shimokawa H, et al. Possible involvement of Rho-kinase in the pathogenesis of hypertension in humans. Hypertension 2001 ; 38 : 1307-10. [Google Scholar]
- Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol 2005 ; 6 : 167-80. [Google Scholar]
- Guilluy C, Bregeon J, Toumaniantz G, et al. The Rho exchange factor Arhgef1 mediates the effects of angiotensin II on vascular tone and blood pressure. Nat Med 2010 ; 16 : 183-90. [Google Scholar]
- Wirth A, Benyo Z, Lukasova M, et al. G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med 2008 ; 14 : 64-8. [Google Scholar]
- Montezano AC, Callera GE, Yogi A, et al. Aldosterone and angiotensin II synergistically stimulate migration in vascular smooth muscle cells through c-Srcregulated redox-sensitive RhoA pathways. Arterioscler Thromb tosc Biol 2008 ; 28 : 1511-8. [Google Scholar]
© 2010 médecine/sciences - Inserm / SRMS
Liste des figures
|
Figure 1 Signalisation de la contraction dans les CMLV. L’augmentation de la phosphorylation des chaînes légères de myosine (P-MLC) induit le développement de la contraction. La stimulation d’un récepteur à sept domaines transmembranaires par un vasoconstricteur produit, via les sous-unités à des protéines G trimériques (Gα) auxquelles ils sont couplés, (1) la stimulation de la kinase des MLC (MLCK) consécutive à l’augmentation du Ca2+ intracellulaire et à sa liaison à la calmoduline (CAM), et (2) l’inhibition de la phosphatase des MLC (MLCP) via la phosphorylation de la MYPT par Rho kinase. GEF : facteurs d’échange de RhoA. |
| Dans le texte | |
|
Figure 2 Rôle essentiel de Arghef1 dans l’HTA dépendante de l’Ang II. A. Effet de l’infusion d’Ang II sur la pression artérielle systolique de souris contrôles (CML-Arhgef1lox/lox) et de souris n’exprimant pas Arhgef1 dans les CML (CMLArhgef1-KO). B. Représentation schématique du rôle respectif de Arhgef12 et Arhgef1 dans l’HTA induite par un excès de sel et l’Ang II (ETAR : récepteur A de l’endothéline; AT1R : récepteur de l’Ang II de type 1). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.