")
")
| Issue |
Med Sci (Paris)
Volume 25, Number 12, Décembre 2009
Anticorps monoclonaux en thérapeutique
|
|
|---|---|---|
| Page(s) | 1011 - 1019 | |
| Section | I - De la conception à la production | |
| DOI | https://doi.org/10.1051/medsci/200925121011 | |
| Published online | 15 décembre 2009 | |
Les anticorps : mieux les connaître pour mieux s’en servir
Antibodies: better knowledge for a better use
1
Laboratoire français du fractionnement et des biotechnologies, Les Ulis, France.
2
Inserm U872, Paris, F-75006 France.
3
Centre de recherche des Cordeliers, Université Pierre et Marie Curie-Paris 6, UMRS 872, Paris, F-75006 France.
4
Université Paris Descartes, UMRS 872, Paris, F-75006 France
5
Inserm U872, Paris, F-75006 France.
6
Équipe 14, Centre de recherche des Cordeliers, Université Pierre et Marie Curie-Paris 6, UMRS 872, Paris, F-75006 France.
7
Équipe 14, Centre de recherche des Cordeliers, 15 rue de l’École de Médecine, 75270 Paris Cedex 06, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
L’utilisation thérapeutique des anticorps monoclonaux connaît une croissance exponentielle. Nos connaissances sur leur structure, en particulier celle des IgG1 largement utilisées en thérapeutique, se sont considérablement accrues. Cependant, de nombreux défis demeurent : l’amélioration de l’efficacité de ces molécules qui reste limitée, la diminution des risques d’événements indésirables chez les patients, et la mise au point de modèles précliniques adéquats, extrapolables à l’être humain. La sélection de cibles pertinentes par rapport à la pathologie visée et aux effets induits par l’anticorps est également un paramètre critique. Face à tous ces défis, la remarquable plasticité moléculaire des anticorps ainsi que les nouvelles possibilités d’ingénierie fondées sur des connaissances accrues de la structure et de la biologie des anticorps et de leurs cibles constituent autant d’espaces de recherche pour faire des anticorps monoclonaux des médicaments exceptionnels pour la santé humaine dans les prochaines années.
Abstract
The therapeutic use of monoclonal antibodies is growing exponentially. Our knowledge on antibody structure, in particular that of IgG1, largely used in the clinic, has progressed remarkably. However, some formidable challenges still remain to be confronted, among which the increase of a yet-limited antibody efficacy, the lowering of the frequency of serious clinical adverse events, and the establishment of pre-clinical models that can be reliably extrapolated to humans represent major goals. The selection of relevant target antigens with regard to the pathology to be treated and to the expected effects of the antibody used is also a critical parameter. Facing these challenges, the amazing molecular plasticity of antibodies, as well as new antibody engineering approaches based on the most recent insights on the structure and biology of antibodies and their targets represent areas of research that will make monoclonal antibodies remarkable drugs for human health in a near future.
© 2009 médecine/sciences - Inserm / SRMS
Un objet complexe mais manipulable
L’utilisation et la manipulation des anticorps (Ac) monoclonaux (Acm) afin d’accroître leur efficacité clinique reposent sur un corpus de connaissances considérable concernant leur structure moléculaire, leur capacité d’interaction avec l’antigène (Ag), ainsi que leur capacité à induire des effets biologiques après la formation d’un complexe avec l’Ag. Les fonctions de fixation à l’Ag et d’induction de fonctions effectrices correspondent à deux entités topologiques distinctes des Ac : la région Fab (fragment antigen binding) qui contient les domaines variables (VH et VL) des chaînes lourdes (H) et légères (L), et la région Fc (fragment cristallisable) [1] (Figure 1). En ce qui concerne les immunoglobulines G (IgG) humaines, qui constituent la classe (ou isotype) de presque tous les Acm à usage thérapeutique actuellement sur le marché, domaines variables et région Fc sont reliés par une région hydrophile et flexible, la région charnière. La flexibilité de cette région, liée à la présence de nombreuses prolines, sérines et thréonines, autorise des torsions allant jusqu’à 180 degrés qui permettent aux Ac de se fixer à des Ag localisés dans différents plans de l’espace [2].
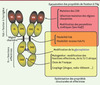 |
Figure 1. Les anticorps sur le métier de l’ingénierie moléculaire. La sélection des CDR (complementarity determining regions), des régions charpentes des domaines VH et VL, l’optimisation des paramètres cinétiques d’interaction permettent d’optimiser la fixation de l’Ac à l’Ag. Le choix de l’isotype, les modifications de la région charnière et des séquences clés impliquées dans les fonctions effectrices, les modifications du profil de glycosylation ou encore le couplage à différents agents cytotoxiques ou stabilisateurs permettent l’optimisation des propriétés effectrices de l’anticorps. |
Interaction épitope-paratope
La zone de l’Ac interagissant avec l’épitope est appelée paratope et est localisée au niveau des domaines VH et VL. Six régions, dites hypervariables, localisées au sein de ces domaines variables et appelées CDR (complementarity determining regions), constituent la zone de contact avec l’épitope et confèrent donc la spécificité de liaison à l’Ag. L’interaction épitope-paratope met en jeu des surfaces d’environ 500 à 600 Å2 [3], le nombre d’acides aminés directement impliqués dans la liaison pouvant aller de six jusqu’à une vingtaine. Cette liaison spécifique de l’épitope à son paratope est non covalente et réversible, et met en jeu des interactions énergétiquement faibles mais nombreuses (forces de van der Waals, forces électrostatiques, ponts hydrogènes et/ou interactions hydrophobes [1, 3]). Selon la nature de l’Ag, le paratope peut être de type cavité (interaction avec des petites molécules), de type sillon (interaction avec des peptides) ou encore de type plan (interaction avec des protéines globulaires). Les Ac sont capables de lier une grande variété d’Ag (sucres, lipides, peptides, composés chimiques, etc.). Cependant, un Ac donné ne reconnaîtra qu’un seul épitope, même si ce dernier peut être partagé par une isoforme, un orthologue ou un Ag différent. Ce partage s’explique soit par la présence de la même séquence d’acides aminés ou d’une séquence très proche, soit par un mimétisme important entre deux molécules qui n’ont structurellement rien à voir l’une avec l’autre. On parle alors de réaction croisée d’un Ac avec d’autres Ag [4, 5].
La règle selon laquelle un Ac ne lie qu’un seul Ag semble cependant relative, au moins avec certains anticorps, comme cela a été montré avec l’étude cristallographique d’un Ac anti-haptène (une petite molécule n’induisant la production d’Ac que lorsqu’elle est couplée à une grosse molécule « porteuse »). Il a été en effet montré que cet Ac pouvait se lier aussi à une protéine, non pas du fait d’un mimétisme moléculaire entre l’épitope de cette protéine et l’haptène, mais du fait de l’existence d’isomères d’anticorps préexistants1 avant toute fixation à l’Ag, ces isomères ayant une capacité de fixation à la protéine ou au haptène [6].
La région Fc contrôle les fonctions effectrices
Les propriétés effectrices de l’Ac sont assurées par la région Fc. Ce sont l’activation de la voie classique du complément (déclenchée par la fixation du C1q) et l’engagement des récepteurs pour la région Fc des Ig (RFc) exprimés par différentes cellules de l’immunité : cellules NK (natural killer), macrophages, polynucléaires, etc. La région Fc joue également un rôle dans la demi-vie des IgG en raison de sa capacité à fixer le récepteur Fc « néonatal » (RFcn) (→) qui participe au recyclage des IgG circulantes [7].
(→) voir C. Magdelaine-Beuzelin et al., p. 1053
Ingénierie des régions Fab et Fc
L’optimisation des Ac passe par une ingénierie manipulant la région Fab (fixation de l’Ac à sa cible) et/ou la région Fc (propriétés effectrices de l’Ac) [8–11] (Figure 1). De nombreux autres travaux ont cherché également à mieux maîtriser la stabilité, le repliement, l’immunogénicité, la demi-vie et la biodisponibilité des Acm ainsi que leurs conditions de production et de purification à une échelle industrielle [8, 9, 12]. Ces différents aspects sont abordés dans d’autres articles de ce numéro.
L’introduction de mutations dirigées ou aléatoires dans les domaines variables permet de moduler l’interaction Ag-Ac afin d’augmenter l’affinité de l’Ac ou de modifier sa spécificité fine [13, 14]. Le choix de ces mutations repose sur la connaissance détaillée des interactions Ag-Ac dans lesquelles les CDR jouent un rôle central. C’est ainsi que la mutagenèse de fragments Fab ou simple chaîne Fv (fragment variable) (single chain Fv, scFv) isolés à partir de banques de phages a permis d’accroître leur affinité d’un facteur 100 à 1 000 [15], cette affinité pouvant atteindre l’ordre du picomolaire. Cependant, comme l’ont montré de nombreuses expériences d’humanisation d’Ac murins, certains acides aminés des régions charpentes des domaines VH et VL, encadrant les CDR, jouent un rôle important dans la conformation finale de ces CDR et dans l’orientation des acides aminés directement impliqués dans l’interaction avec l’Ag [4]. Des mutations affectant ces acides aminés peuvent conduire à des modifications considérables de l’affinité de l’Ac pour l’Ag [13, 15].
L’affinité d’un Ac s’apprécie par la mesure des constantes d’équilibre d’association (KA) et de dissociation (KD) du complexe Ag-Ac. Le KD correspond à la concentration molaire de l’Ag nécessaire à l’occupation de la moitié des molécules d’Ac disponibles en solution et est la constante la plus souvent mesurée pour déterminer l’affinité d’un Ac. Les constantes cinétiques de formation et de dissociation du complexe Ag-Ac, appelées respectivement kon et koff, sont mesurées afin de déterminer la vitesse d’atteinte de l’équilibre ainsi que le temps de fixation de l’Ac à son Ag. Ces paramètres cinétiques sont très importants pour les fonctions de l’Ac. Depuis l’autorisation de mise sur le marché (AMM) du rituximab, un Acm anti-CD20 initialement utilisé dans le traitement des lymphomes B non hodgkiniens [16], les Acm anti-CD20 ont fait l’objet de nombreux travaux visant à comprendre leurs mécanismes d’action (→). L’un de ces travaux a montré que les Acm présentant un temps de fixation au CD20 prolongé recrutent plus efficacement le C1q, induisant ainsi une cytotoxicité dépendante du complément (complement-dependent cytotoxicity, CDC) plus élevée [17]. La sélection de molécules aux constantes cinétiques optimisées est donc un moyen efficace pour améliorer les propriétés fonctionnelles d’un Acm.
(→) voir G. Cartron et J.F. Rossi, p. 1085
Le choix et la manipulation de la région Fc sont également essentiels pour disposer d’un Acm capable d’induire les fonctions effectrices désirées. La plupart des Acm actuellement sur le marché (16/26), qu’ils soient chimériques (contenant des VH et des VL d’origine murine et des régions constantes humaines), humanisés (dont les VH et les VL ont une région charpente humaine et des CDR murins, les régions constantes étant humaines), ou totalement humains, sont des IgG1 : isotype capable d’activer la voie classique du complément et de fixer les RFc pour les IgG (RFcγ). Dans trois cas seulement, une IgG2 et deux IgG4 ont été choisies afin de ne pas activer ces fonctions qui peuvent être à l’origine d’événements indésirables comme la production massive et brutale de cytokines [18].
Des techniques moléculaires ont été développées pour modifier la région Fc des IgG1 afin d’optimiser leur activation de la voie classique du complément [19] (les acides aminés jouant un rôle crucial dans la fixation du Fc au C1q étant l’acide aspartique 270 et la proline 329 [20]) ou engager les RFcγ activateurs (notamment le RFcγIII/CD16) [21, 22] (les acides aminés directement impliqués dans le site de fixation à ce récepteur étant les acides aminés 234 à 238 localisés dans la partie carboxy-terminale de la région charnière [23]), conduisant à une cytotoxicité dépendante des Ac (antibody-dependent cell cytotoxicity, ADCC) accrue. Ces techniques incluent la mutagenèse de la région Fc [21, 22] et la sélection de glycoformes particulières, notamment les glycoformes dépourvues de, ou ayant un faible taux de fucose [24–26]. Les IgG1 humaines présentent en effet un site de N-glycosylation au niveau de l’asparagine 297 auquel est fixé un carbohydrate incluant des N-acétylglucosamines, du mannose et, éventuellement, du galactose, du fucose et des acides sialiques (→). Du fait de l’existence chez l’homme de RFcγ inhibiteurs (RFcγIIB/CD32) qui pourraient réduire l’efficacité anti-tumorale d’Acm à usage thérapeutique [27], des Acm obtenus par mutagenèse fixant plus faiblement ces RFcγII inhibiteurs tout en conservant une bonne fixation, voire ayant une fixation accrue aux RFcγ activateurs, sont actuellement étudiés [21, 22].
(→) voir A. Beck et al., page 1024
Le drôle de jeu des épitopes et… des paratopes
Le choix d’un Ac à usage thérapeutique est également dicté par l’Ag lui-même et l’épitope reconnu [28], ses caractéristiques structurales et fonctionnelles, ainsi que son rôle dans la pathologie ciblée. Concevoir les interactions épitope-paratope comme un système rigide est réducteur. Des degrés de liberté existent, les Ac étant des molécules flexibles du fait de leur région charnière et des ajustements de la conformation de leurs domaines variables lors de l’interaction avec l’Ag. La structure de la région charnière permet de réguler la mobilité Fab-Fab et l’espacement Fab-Fc. Il n’est donc pas surprenant que l’isotype IgG1 soit le plus représenté parmi les Acm utilisés en oncologie car sa région charnière offre le meilleur compromis entre flexibilité des régions Fab et disponibilité de la région Fc. Les réarrangements consécutifs à la fixation de l’Ag sont également le reflet du caractère plastique des Ac. La mise en évidence de phénomènes de réarrangements induits (induced fit) lors de la fixation à l’Ag (principalement dans le cas de protéines) a remis en cause la vision figée des interactions Ag-Ac [29]. Lorsqu’un Ac lie son Ag pour former un complexe stable du point de vue cinétique et thermodynamique, de fins réarrangements structuraux du paratope, induits par la liaison à l’épitope, se produisent du fait de la flexibilité des chaînes latérales de certains acides aminés des régions charpentes. L’épitope peut alors également subir de fins remaniements structuraux. Ces ajustements induits renforcent l’interaction entre les partenaires dans une dynamique d’interactions bidirectionnelles.
Cette plasticité des domaines variables a-t-elle un impact sur les fonctions effectrices d’un Ac ? L’ingénierie des anticorps anti-CD20, passés d’une version d’anticorps recombinants de première génération à des versions de seconde génération correspondant au développement de formats plus affins, et/ou reconnaissant des épitopes différents et/ou présentant des propriétés fonctionnelles « optimisées » (ADCC, CDC, apoptose) [30], a jeté une lumière nouvelle sur cette question, grâce à l’étude menée par Umana et al., portant sur un nouvel Acm anti-CD20, le GA101 [31]. Les anticorps anti-CD20 se classent en types I ou II selon leurs caractéristiques effectrices in vitro [31–33] (→). Les Acm de type I induisent la relocalisation de CD20 dans les radeaux lipidiques et une forte CDC, mais provoquent une faible apoptose. Inversement, les Acm de type II induisent une forte apoptose et une faible CDC, mais ne provoquent pas de relocalisation dans les radeaux lipidiques. Dans leur étude, Umana et al. décrivent le GA101, version humanisée d’un Acm anti-CD20 murin de type I. Lors du processus d’humanisation, une modification ponctuelle de la région charpente d’un des domaines variables a été réalisée. Cette modification a converti le GA101 en Acm de type II, augmentant ainsi son potentiel proapoptotique et diminuant ses capacités à induire la CDC et à réorganiser le CD20 à la surface cellulaire [31]. Cette modification très importante des propriétés effectrices provoquée par une unique modification des régions charpentes des domaines variables souligne l’impact fonctionnel que peut avoir la plasticité structurale des Ac et la possibilité d’utiliser cette plasticité pour le développement d’Ac optimisés.
(→) voir G. Cartron et J.F. Rossi, page 1085
Les cibles : bonnes ou mauvaises, il faut savoir choisir…
Parmi les paramètres dont il faut tenir compte pour développer un Acm à usage thérapeutique, le choix de la molécule cible est majeur. Si cette cible peut s’imposer d’elle-même dans certaines pathologies sur la base de travaux scientifiques et cliniques, son choix relève néanmoins toujours d’un compromis délicat entre efficacité clinique, éventuels effets secondaires, indications, propriété intellectuelle, marché potentiel et cohérence avec la stratégie industrielle de l’entreprise concernée. Nous nous limiterons ici à décrire quelques aspects structuraux et fonctionnels concernant les caractéristiques des cibles actuellement étudiées visant à l’obtention d’une bonne efficacité de l’Acm sans induction d’événements indésirables graves.
Acm ciblant des cytokines
Sur les 26 anticorps ayant obtenu une AMM [9, 34], un peu moins d’un quart a pour cible une molécule soluble : cytokine [tumor necrosis factor-α (TNFα), chaîne commune α de l’interleukine 12 (IL-12) et de l’IL-23], Ac (IgE), composant du complément (C5) ou facteur de croissance (vascular endothelial growth factor, VEGF). En raison de leurs effets pléiotropes et de nombreuses redondances dans leurs effets biologiques, les cytokines ne semblaient pas destinées à devenir des cibles pour les Acm à usage thérapeutique. Cependant, la démonstration du rôle central joué par le TNFα dans la physio-pathologie de désordres immunitaires chez l’homme a conduit au développement d’Acm anti-TNFα (infliximab, adalimumab, certolizumab pégol) (→). En dépit de l’énorme avancée des connaissances sur la biologie des cytokines, le TNFα est resté jusqu’à fin 2008 la seule cytokine pour laquelle des Acm ont reçu une AMM (l’ustékinumab anti-IL-12/23 ayant alors reçu une AMM [35]), même si le ciblage d’autres cytokines [IFN (interféron)-γ, IL-8…] [36, 37] par des Acm neutralisant leurs interactions avec les récepteurs spécifiques est actuellement évalué dans des essais cliniques.
(→) voir L. Semerano et M.C. Boissier, page 1108 ; J. Sibilia, p. 1033
La compréhension des effets induits par le ciblage d’une cytokine par un Acm peut être d’autant plus difficile que la présence des molécules ciblées peut se détecter également à la membrane des cellules. L’exemple du TNFα dans le contexte de la polyarthrite rhumatoïde (PR) illustre bien cette complexité (→). Le TNFα soluble (sTNFα) joue un rôle central dans la physiopathologie de la PR et l’utilisation des anti-TNFα a révolutionné la prise en charge des patients. Cependant, certains individus traités par anti-TNFα présentent une vulnérabilité aux infections, en particulier à la tuberculose (→). Cet effet secondaire a été expliqué par l’existence de TNFα membranaire (mTNFα) à la surface de lymphocytes T qui est responsable de l’activation des monocytes producteurs de TNFα soluble (sTNFα) par les lymphocytes T. Son blocage par les Acm anti-TNFα conduit à une diminution importante de la production de sTNFα [38] qui est un facteur de survie des lymphocytes T effecteurs/mémoires impliqués dans la défense anti-tuberculose [39]. Les Acm anti-TNFα agiraient donc en inhibant la survie des lymphocytes T effecteurs/mémoires spécifiques induite par le sTNFα, favorisant une réactivation de l’infection latente. Un travail récent a par ailleurs montré que les lymphocytes T effecteurs-mémoires CD8+CCR7-CD45RA+ responsables d’une forte activité anti-microbienne contre M. tuberculosis exprimaient fortement du mTNFα et pouvaient être lysés par une réaction de CDC faisant suite à la fixation de l’Acm anti-TNFα infliximab au mTNFα que ces cellules expriment à leur surface [40].
(→) voir L. Semerano et M.C. Boissier, p. 1108
(→) voir E. Rigal et al., page 1135
Acm ciblant des antigènes membranaires
La plupart des cibles des Acm qui ont une AMM ou qui sont dans une phase avancée de développement clinique sont des Ag membranaires (Tableau I) [9, 34]. L’effet de l’Acm peut être dû à des mécanismes effecteurs aboutissant à la mort de la cellule cible et/ou à l’induction de mécanismes moléculaires intracellulaires après interaction de l’Ac avec la molécule cible (apoptose, cytostase) (Figure 2). L’expression et/ou le comportement de la cible membranaire, en termes de densité, de trafic et/ou de signalisation après interaction avec l’Acm, sont des facteurs déterminants de l’efficacité de ce dernier. Un mécanisme additionnel qui permet à la cellule cible d’échapper à la cytotoxicité dépendante du complément est l’expression à la surface de cette cellule de molécules inhibitrices de l’action du complément [membrane cofactor protein (MCP)/CD46, decay accelerating factor (DAF)/CD55, CD59] (→).
Exemples d’anticorps ou de fragments d’anticorps en cours de développement clinique en oncologie.
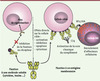 |
Figure2. Les anticorps à l’assaut des antigènes solubles ou membranaires. L’action d’un Ac varie selon sa cible, selon que celle-ci est soluble (à gauche) ou membranaire (à droite). Dans la première situation, l’Ac empêche la fixation de l’Ag (cytokine, chimiokine, toxine…) à son récepteur présent à la surface des cellules, ce qui conduit à l’absence de toute signalisation normalement induite par cet antigène. De façon similaire, des Acm dirigés contre la protéine d’enveloppe d’un virus responsable de la fixation du virion à un récepteur cellulaire vont inhiber cette fixation et bloquer ainsi l’entrée du virus. Dans la deuxième situation, l’Ac va fixer l’Ag présent à la surface de la cellule cible, induisant ainsi soit des effets directs (activation ou inhibition de l’activation cellulaire, apoptose, cytostase) soit le recrutement et l’activation d’effecteurs cellulaires (cellules NK, monocytes, polynucléaires neutrophiles…) ou moléculaires (activation de la voie classique du complément après fixation de la molécule C1q), sans que ces deux types d’effets soient mutuellement exclusifs. |
(→) voir G. Socié et al., page 1126
Les cibles membranaires des Acm actuellement sur le marché sont des récepteurs de facteurs de croissance - epidermal growth factor-receptor (EGF-R), human epidermal growth factor receptor-2/neu (HER-2/neu), même si dans ce dernier cas il n’y a pas de ligand connu - ou de cytokine (IL-2-Rα/CD25), des molécules d’adhérence impliquées dans les interactions cellule-cellule (epithelial cell adhesion molecule, Ep-CAM ; chaîne a4 du very late antigen-4, VLA-4 ; molécule CD11a, sous-unité du leukocyte function-associated antigen, LFA-1) ou des protéines transmembranaires dont la fonction n’est pas totalement élucidée (CD20, CAMPATH-1/CD52, CD33). De nouvelles cibles sont en cours d’évaluation, notamment des récepteurs : insulin-like growth factor 1 receptor (IGF1-R)/CD221 (→), chemokine (C-C motif) receptor 4 (CCR4)/CD194, IL-5-Rα/CD125), et des molécules impliquées dans le contrôle positif ou négatif de l’activation cellulaire : cytotoxic T-lymphocyte antigen 4 (CTLA-4)/CD152, CD19, CD22, CD30, receptor activator of nuclear factor kappa ligand (RANK-L)/CD254… (Tableau I).
(→) voir A. Bodmer et al., page 1090
L’obstacle de la modulation antigénique
La variation d’expression des molécules membranaires constitue un obstacle majeur au succès des thérapies chez l’homme. Cette variation d’expression peut être liée à la biologie de l’Ag cible et/ou être la conséquence de la fixation de l’Ac sur son Ag provoquant son internalisation (Figure 3). La faible efficacité du rituximab dans la leucémie lymphoïde chronique (LLC) a été corrélée à la faible densité des molécules CD20 à la surface des cellules tumorales, un problème qui semble avoir été résolu grâce à l’utilisation d’Ac optimisés. C’est ainsi qu’en collaboration avec le Laboratoire français du fractionnement et des biotechnologies (LFB), notre équipe a montré qu’un Ac anti-CD20 optimisé pour la fixation au RFcγIIIA par modification de la composition en sucre de sa région Fc présente in vitro des capacités pro-apopto-tiques et de CDC équivalentes à celles du rituximab mais une capacité d’ADCC fortement accrue [41] (→). Cette différence d’activité lytique in vitro est d’autant plus prononcée que la densité d’Ag est plus faible, comme dans le cas de la LLC. Cette observation démontre que l’ingénierie moléculaire des Ac peut apporter des solutions au problème posé par la faible expression de l’Ag à la surface des cellules cibles. Le phénomène de modulation antigénique peut cependant être exploité favorablement à des fins thérapeutiques. L’omalizumab, un Acm anti-IgE (Fc) indiqué pour le traitement de l’asthme allergique, a un double effet conduisant à une diminution des réactions d’hypersensibilité immédiate. Il se fixe au domaine Cε3 de la région Fc des IgE circulantes, bloquant leur interaction avec le RFc de haute affinité pour les IgE (RFcεI), et induit alors une baisse d’expression du RFcεI à la surface des basophiles et des mastocytes [42, 43].
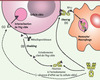 |
Figure 3. Évasion des cellules cibles de l’action des anticorps par modulation de l’expression de l’Ag. La diminution de l’expression de l’Ag à la surface de la cellule cible permet à celle-ci de résister à l’action des Ac. Selon l’Ag considéré, cette modulation pourra se traduire par une internalisation de l’Ag (1), un clivage des ectodomaines de l’Ag (shedding) (2) mettant en jeu des métalloprotéinases et/ou par un « arrachement » (shaving) (3) du complexe Ac-Ag assuré par les monocytes/macrophages par l’intermédiaire des RFcγ. |
(→) voir R. Urbain et al., page 1141
La modulation antigénique peut être aussi la conséquence de deux autres mécanismes : le shedding et le shaving (Figure 3). Le shedding est un mécanisme de clivage des protéines membranaires. Ce processus implique une protéolyse limitée des ectodomaines des protéines transmembranaires par des métalloprotéinases [44]. Les molécules CD20, Campath-1/CD52 et CD33, cibles d’Acm actuellement sur le marché, sont soumises à ce mécanisme [45, 46], limitant potentiellement l’effet thérapeutique in vivo des Acm correspondants. Le shaving résulte de la capacité des monocytes/macrophages à fixer puis « arracher » de la membrane des cellules cibles les complexes molécule cible-Acm et à les internaliser. Ce processus, dépendant des RFcγ, a été impliqué dans la diminution de l’expression de CD20 observée à la surface de cellules leucémiques chez des patients présentant une LLC et traités par le rituximab [30, 47].
La prise en compte du rapport bénéfice-risque
La sélection d’une cible doit évidemment tenir compte du rapport bénéfice/risque. La plupart des Acm occasionnent des effets secondaires (vomissements, maux de tête, nausées, etc.) le plus souvent sans conséquences graves, ce qui fait de l’utilisation des Acm en clinique une thérapie assez sûre et bien tolérée par rapport à d’autres, notamment en oncologie. Cependant, des effets secondaires importants ont été rapportés. Le trastuzumab (anti-HER-2/neu ou c-erbB-2), indiqué dans le cancer du sein métastatique, présente une cardiotoxicité importante dans environ 10 % des cas [48]. Cette toxicité s’explique par l’expression d’erbB-2 à la surface des cardiomyocytes. Par contre, un autre Acm anti-erbB-2, le pertuzumab, qui bloque l’hétéro-dimérisation d’erbB-2, actuellement en essai clinique, n’a pas montré de cardiotoxicité à l’issue d’un essai de phase I [49], suggérant que le choix de l’épitope pour un même Ag peut jouer un rôle crucial dans les effets des Acm. Un autre exemple illustrant les difficultés rencontrées avec certaines cibles est celui de l’Acm immuno-modulateur anti-CD28 TGN1412, un superagoniste capable d’induire in vivo chez l’animal l’activation et l’expansion de lymphocytes T régulateurs et susceptible d’améliorer le traitement de maladies auto-immunes [50]. Malgré des études précliniques démontrant une toxicité acceptable et une bonne efficacité dans différents modèles animaux, un essai clinique de phase I sur six volontaires sains a tourné au drame. Moins d’une heure après la première infusion, les six volontaires ont développé un syndrome très sévère dû à une production massive de cytokines (cytokine storm syndrome), nécessitant leur transfert en réanimation (→). Cet incident très grave a souligné l’importance de la compréhension des mécanismes biologiques liés à l’immunomodulation (CD28 est définie comme une molécule de costimulation exprimée de façon constitutive à la surface de tous les lymphocytes CD4+), et a montré les limites des modèles animaux, conduisant à redéfinir les critères de sécurité relatifs aux premières injections chez l’homme [51]. Malgré ces difficultés d’appréciation du risque, un nombre croissant d’Acm à vocation immunomodulatrice est actuellement développé, que ce soit pour des traitements de maladies auto-immunes et inflammatoires ou pour des traitements anticancéreux (anti-RANK-L/CD254, anti-CD80, anti-CTLA-4/CD152) (→).
(→) voir M. Pallardy, page 1130
(→) voir A. Bodmer et al., page 1090
Conclusion
Les Ac comptent parmi les molécules de l’immunité les mieux connues. Leur utilisation en clinique humaine connaît une expansion rapide, malgré les revers qui ont jalonné leur histoire. La connaissance toujours plus précise de ces molécules, leur ingénierie toujours plus sophistiquée et nos connaissances accrues de leurs fonctions en font des candidats remarquables pour le développement de nouvelles thérapies. Cependant, le succès in fine d’un Acm particulier dépend d’un équilibre instable entre différents paramètres :
pertinence clinique (rôle clé joué par la cellule ou la molécule ciblée par l’Ac dans la pathologie indiquée) ;
propriétés structurales et fonctionnelles de l’Ac (affinité, stabilité, propriétés effectrices telles que l’ADCC ou la CDC) et de la molécule cible (densité, accessibilité, internalisation, libération sous forme soluble, spécificité d’expression tissulaire, pléiotropie des effets, redondance d’effets avec d’autres molécules) ;
choix de l’épitope (permettant ou non une compétition avec un ligand, induisant un mécanisme de cytostase ou de mort cellulaire après fixation de l’Ac)
gestion raisonnée des essais précliniques (choix du modèle animal, évaluation de l’efficacité et des effets secondaires) et cliniques (sélection des patients inclus fondée aussi sur des paramètres liés à l’utilisation de l’Ac et/ou à la cible moléculaire ; suivi de paramètres biologiques liés à l’utilisation d’un Acm).
Les prochaines années verront certainement l’éclosion d’une nouvelle génération d’Acm ayant des structures optimisées pour des fonctions particulières [ADCC, CDC, demi-vie sérique (T1/2)], ainsi qu’un retour sur le devant de la scène de formats étudiés dans les années 1980 (Ac bispécifiques (→), Ac conjugués) et la montée en puissance de miniformats [VH/VL, single chain Fv, domaine VH simple de la chaîne H des camélidés (VHH)] (→). Nos connaissances accrues de la structure et du rôle de nombreuses molécules cibles, notamment dans le domaine de l’oncologie et des protéines solubles (toxines, cytokines) vont aussi renforcer les approches fondées sur une utilisation combinée de plusieurs Acm, dirigés contre des molécules différentes ayant des fonctions différentes ou contre des épitopes différents de la même molécule. Ces approches « oligoclonales » permettront certainement à terme d’accroître l’efficacité thérapeutique des Ac, une sorte de sérothérapie moderne, une idée chère à Emil von Behring et Shibasaburo Kitasato (→) [52].
(→) voir A. Pèlegrin et B. Robert, page 1155
(→) voir P. Chames et D. Baty, page 1159
(→) voir H. Watier, page 999
Conflit D’Intérêts
Les auteurs déclarent n’avoir aucun conflit d’intérêts concernant les données publiées dans cet article.
On désigne sous ce terme le fait qu’« une même molécule d’anticorps peut préexister sous plusieurs conformations structurales, chacune étant susceptible de reconnaître un antigène spécifique. Un équilibre existe entre plusieurs (au moins deux) conformations structurales (isomères) d’un anticorps unique, chaque isomère étant capable d’interagir spécifiquement avec des antigènes aussi différents qu’une petite molécule chimique (haptène) et une protéine globulaire » (extrait de [53]).
Références
- Amzel LM, Poljak RJ. Three-dimensional structure of immunoglobulins. Annu Rev Biochem 1979; 48 : 961–97. [Google Scholar]
- Saphire EO, Stanfield RL, Crispin MD, et al. Contrasting IgG structures reveal extreme asymetry and flexibility. J Mol Biol 2002; 319 : 9–18. [Google Scholar]
- Mariuzza RA, Phillips SE, Poljack RJ. The structural basis of antigen-antibody recognition. Annu Rev Biophys Biophys Chem 1987; 16 : 139–59. [Google Scholar]
- Mian IS, Bradwell AR, Olson AJ. Structure, function and properties of antibody binding sites. J Mol Biol 1991; 217 : 133–51. [Google Scholar]
- Oldstone MB. Molecular mimicry and immune-mediated diseases. FASEB J 1998; 12 : 1255–65. [Google Scholar]
- James LC, Roversi P, Tawfik DS. Antibody multispecificity mediated by conformational diversity. Science 2003; 299 : 1362–7. [Google Scholar]
- Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol 2007; 7 : 715–25. [Google Scholar]
- Carter P. Potent antibody therapeutics by design. Nat Rev Immunol 2006; 6 : 343–57. [Google Scholar]
- Reichert JM. Monoclonal antibodies as innovative therapeutics. Curr Pharm Biotechnol 2008; 9 : 423–30. [Google Scholar]
- Teillaud JL. Engineering of monoclonal antibodies and antibody-based fusion proteins: successes and challenges. Exp Opin Biol Ther 2005; 5 : S1–13. [Google Scholar]
- Winter G, Milstein C. Man-made antibodies. Nature 1991; 349 : 293–9. [Google Scholar]
- Bourel D, Teillaud JL. Anticorps monoclonaux : tours et détours technologiques pour de nouveaux espoirs thérapeutiques. CR Biol 2006; 329 : 217–27. [Google Scholar]
- Hawkins RE, Russell SJ, Winter G. Selection of phage antibodies by binding affinity. Mimicking affinity maturation. J Mol Biol 1992;226 : 889–96. [Google Scholar]
- Coulon S, Pellequer JL, Blachère T, et al. Functional characterization of an anti-estradiol antibody by site-directed mutagenesis and molecular modelling: modulation of binding properties and prominent role of the V(L) domain in estradiol recognition. J Mol Recognit 2002; 15 : 6–18. [Google Scholar]
- Schier R, McCall A, Adams GP, et al. Isolation of picomolar affinity anti-c-erbB-2 single-chain Fv by molecular evolution of the complementarity determining regions in the center of the antibody binding site. J Mol Biol 1996; 263 : 551–67. [Google Scholar]
- McLaughlin P, Grillo-López AJ, Link BK, et al. Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four-dose treatment program. J Clin Oncol 1998; 16 : 2825–33. [Google Scholar]
- Teeling JL, French RR, Cragg MS, et al. Characterization of new human CD20 monoclonal antibodies with potent cytolytic activity against non-Hodgkin lymphomas. Blood 2004; 104 : 1793–800. [Google Scholar]
- Jeyarajah DR, Thistlethwaite JR. General aspects of cytokine-release syndrome: timing and incidence of symptoms. Transplant Proc 1993; 25 : 16–20. [Google Scholar]
- Idusogie EE, Wong PY, Presta LG, et al. Engineered antibodies with increased activity to recruit complement. J Immunol 2001; 166 : 2571–5. [Google Scholar]
- Idusogie EE, Presta LG, Gazzano-Santoro H, et al. Mapping of the C1q binding site on Rituxan, a chimeric antibody with a human IgG1 Fc.J Immunol 2000; 164 : 4178–84. [Google Scholar]
- Shields RL, Namenuk AK, Hong K,et al. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R J Biol Chem 2001; 276 : 6591–604. [Google Scholar]
- Lazar GA, Dang W, Karki S, et al. Engineered antibody Fc variants with enhanced effector function. Proc Natl Acad Sci USA 2006; 103 : 4005–10. [Google Scholar]
- Sibéril S, Dutertre CA, Boix C, et al. Molecular aspects of human FcgammaR interactions with IgG: functional and therapeutic consequences. Immunol Lett 2006; 106 : 111–8. [Google Scholar]
- Shields RL, Lai J, Keck R,et al. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fc gamma RIII and antibody-dependent cellular toxicity. J Biol Chem 2002; 277 : 26733–40. [Google Scholar]
- Shinkawa T, Nakamura K, Yamane N, et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J Biol Chem 2003; 278 : 3466–73. [Google Scholar]
- Sibéril S, de Romeuf C, Bihoreau N, et al. Selection of a human anti-RhD monoclonal antibody for therapeutic use: impact of IgG glycosylation on activating and inhibitory Fc gamma R functions. Clin Immunol 2006; 118 : 170–9. [Google Scholar]
- Clynes RA, Towers TL, Presta LG., et al. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat Med 2000; 6 : 443–6. [Google Scholar]
- Cragg MS, Glennie MJ. Antibody specificity controls in vivo effector mechanisms of anti-CD20 reagents. Blood 2004; 103 : 2738–43. [Google Scholar]
- Davies DR, Cohen GH. Interactions of protein antigens with antibodies. Proc Natl Acad Sci USA 1996; 93 : 7–12. [Google Scholar]
- Taylor R, Lindorfer MA. Immunotherapeutic mechanisms of anti-CD20 monoclonal antibodies. Curr Opinion Immunol 2008; 20 : 444–9. [Google Scholar]
- Umana P, Moessner E, Bruenker P, et al. Novel 3rd generation humanized type II CD20 antibody with glycoengineered Fc and modified elbow hinge for enhanced ADCC and superior apoptosis induction. ASH Annual Meeting Abstracts 2006; 108 : 229. [Google Scholar]
- Chan HT, Hughes D, French RR, et al. CD20-induced lymphoma cell death is independent of both caspases and its redistribution into Triton-X100 insoluble membrane rafts. Cancer Res 2003; 63 : 5480–9. [Google Scholar]
- Cragg MS, Morgan SM, Chan HT, et al. Complement-mediated lysis by anti-CD20 mAb correlates with segregation into lipid rafts. Blood 2003; 101 : 1045–52. [Google Scholar]
- Weiner L, Dhodapkar MV, Ferrone S. Monoclonal antibodies for cancer immunotherapy. Lancet 2009; 373 : 1033–40. [Google Scholar]
- Gottlieb A, Menter A, Mendelsohn A, et al. Ustekinumab, a human interleukin 12/23 monoclonal antibody, for psoriatic arthritis: randomized, double-blind, placebo-controlled, crossover trial. Lancet 2009; 373 : 633–40. [Google Scholar]
- Hommes DW, Mikhajlova TL, Stoinov Z, et al. Fontolizumab, a humanized anti-interferon γ antibody, demonstrates safety and clinical activity in patients with moderate to severe Crohn’s disease. Gut 2006; 55 : 1131–7. [Google Scholar]
- Skov L, beurskens FJ, Zachariae COC, et al. IL-8 as antibody therapeutic target in inflammatory diseases: reduction of clinical activity in palmoplantar pustulosis. J Immunol 2008; 181 : 669–79. [Google Scholar]
- Rossol M, Meusch U, Pierer M,et al. Interaction between transmembrane TNF and TNFR1/2 mediates the activation of monocytes by contact with T cells. J Immunol 2007; 179 : 4239–48. [Google Scholar]
- Emilie D. Effect on TNF antagonists on the T-lymphocyte response. Joint Bone Spine 2007;74 : 558–9. [Google Scholar]
- Bruns H, Meinken C, Schauenberg P,et al. Anti-TNF immunotherapy reduces CD8+ T cell-mediated antimicrobial activity against mycobacterium tuberculosis in humans. J Clin Invest 2009; 119 : 1167–77. [Google Scholar]
- De Romeuf C, Dutertre CA, Le Garff-Tavernier M, et al. Chronic lymphocytic leukemia cells are efficiently killed by an anti-CD20 monoclonal antibody selected for improved engagement of FcγRIIIA/CD16. Br J Haematol 2008; 140 : 635–43. [Google Scholar]
- Lin H, Boesel KM, Griffith DT, et al. Omalizumab rapidly decreases nasal allergic response and FcepsilonRI on basophils. J Allergy Clin Immunol 2004; 113 : 297–302. [Google Scholar]
- Ames SA, Gleeson CD, Kirkpatrick P. Omalizumab. Nat Rev Drug Disc 2004; 3 : 199–204. [Google Scholar]
- Arribas J, Borroto A. Protein ectodomain shedding. Chem Rev 2002; 102 : 4627–38. [Google Scholar]
- Giles FJ, Vose JM, Do KA, et al. Circulating CD20 and CD52 in patients with non-Hodgkin’s disease. Br J Haematol 2003; 123 : 850–7. [Google Scholar]
- Bidermann B, Gil D, Bowen DT, et al. Analysis of the CD33-related siglec family reveals that siglec-9 is an endocytic receptor expressed on subsets of acute myeloid leukemia cells and absent from normal hematopoietic progenitors. Leuk Res 2007; 31 : 211–20. [Google Scholar]
- Beum PV, Kennedy AD, Williams ME, et al. The shaving reaction: rituximab/CD20 complexes are removed from mantle cell lymphoma and chronic lymphocytic leukemia cells by THP-1 monocytes. J Immunol 2006; 176 : 2600–9. [Google Scholar]
- Klastersky J. Adverse effects of the humanized antibodies used as cancer therapeutics. Curr Opin Oncol 2006; 18 : 316–20. [Google Scholar]
- Agus DB, Gordon MS, Taylor C. Phase I clinical study of pertuzumab, a novel HER dimerization inhibitor, in patients with advanced cancer. J Clin Oncol 2005; 23 : 2534–43. [Google Scholar]
- Schraven B, Kalinke U. CD28 superagonists: what makes the difference in humans ? Immunity 2008; 28 : 591–5. [Google Scholar]
- Loisel S, Ohresser M, Pallardy M, et al. Relevance, advantages and limitations of animal models used in the development of monoclonal antibodies for cancer treatment. Crit Rev Oncol Hematol 2007;62 : 34–42. [Google Scholar]
- Behring E, Kitasato S. Über das Zustandekommen der Diphterie-Immunität und der Tetanus-Immunität bei Thieren. Dtsch Med Wochenschr 1890; 16 : 1113–4. [Google Scholar]
- Ducancel F. Quand la diversité du répertoire des immunoglobulines augmente ! Med Sci (Paris) 2003; 19 : 790–2. [Google Scholar]
Liste des tableaux
Exemples d’anticorps ou de fragments d’anticorps en cours de développement clinique en oncologie.
Liste des figures
|
Figure 1. Les anticorps sur le métier de l’ingénierie moléculaire. La sélection des CDR (complementarity determining regions), des régions charpentes des domaines VH et VL, l’optimisation des paramètres cinétiques d’interaction permettent d’optimiser la fixation de l’Ac à l’Ag. Le choix de l’isotype, les modifications de la région charnière et des séquences clés impliquées dans les fonctions effectrices, les modifications du profil de glycosylation ou encore le couplage à différents agents cytotoxiques ou stabilisateurs permettent l’optimisation des propriétés effectrices de l’anticorps. |
| Dans le texte | |
|
Figure2. Les anticorps à l’assaut des antigènes solubles ou membranaires. L’action d’un Ac varie selon sa cible, selon que celle-ci est soluble (à gauche) ou membranaire (à droite). Dans la première situation, l’Ac empêche la fixation de l’Ag (cytokine, chimiokine, toxine…) à son récepteur présent à la surface des cellules, ce qui conduit à l’absence de toute signalisation normalement induite par cet antigène. De façon similaire, des Acm dirigés contre la protéine d’enveloppe d’un virus responsable de la fixation du virion à un récepteur cellulaire vont inhiber cette fixation et bloquer ainsi l’entrée du virus. Dans la deuxième situation, l’Ac va fixer l’Ag présent à la surface de la cellule cible, induisant ainsi soit des effets directs (activation ou inhibition de l’activation cellulaire, apoptose, cytostase) soit le recrutement et l’activation d’effecteurs cellulaires (cellules NK, monocytes, polynucléaires neutrophiles…) ou moléculaires (activation de la voie classique du complément après fixation de la molécule C1q), sans que ces deux types d’effets soient mutuellement exclusifs. |
| Dans le texte | |
|
Figure 3. Évasion des cellules cibles de l’action des anticorps par modulation de l’expression de l’Ag. La diminution de l’expression de l’Ag à la surface de la cellule cible permet à celle-ci de résister à l’action des Ac. Selon l’Ag considéré, cette modulation pourra se traduire par une internalisation de l’Ag (1), un clivage des ectodomaines de l’Ag (shedding) (2) mettant en jeu des métalloprotéinases et/ou par un « arrachement » (shaving) (3) du complexe Ac-Ag assuré par les monocytes/macrophages par l’intermédiaire des RFcγ. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.