")
")
| Issue |
Med Sci (Paris)
Volume 25, Number 10, Octobre 2009
Accidents vasculaires cérébraux
|
|
|---|---|---|
| Page(s) | 855 - 857 | |
| Section | M/S revues : Accidents vasculaires cérébraux (2) | |
| DOI | https://doi.org/10.1051/medsci/20092510855 | |
| Published online | 15 octobre 2009 | |
Comment optimiser l’utilisation du tPA ?
How to neutralize the neurotoxic effects of tPA
Inserm U919, Sérine protéases et physiopathologie de l’unité neurovasculaire, UMR-CNRS 6232 CINAPS, Cyceron, F-14074 Caen, France
Université de Caen Basse-Normandie, GIP Cycéron, boulevard Henri Becquerel, 14074 Caen, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Les options thérapeutiques associées à la thrombolyse
L’administration intraveineuse ou intra-artérielle de l’activateur tissulaire du plasminogène (tPA, Figure 1) agissant comme agent thrombolytique est confrontée à deux écueils : comment rendre son utilisation plus sûre et/ou efficace si l’administration de tPA intervient plus de 3 ou 4 heures après le début des symptômes ? De même se pose la question de l’utilisation conjointe de la thrombolyse et des ultrasons ou de l’association de la thrombolyse avec l’hypothermie, deux options qui font aujourd’hui l’objet d’essais cliniques. On peut aussi envisager une voie de recherche thérapeutique ciblant la cascade d’événements biochimiques et physiologiques qui suit un épisode ischémique, en parallèle des stratégies de reperfusion. En dépit de l’échec récent des essais cliniques SAINT II (stroke acute ischemic NXY-059 trial) qui examinaient l’effet du tPA en combinaison avec le NXY-0591, des thérapies combinées avec le tPA devraient être encore considérées et encouragées.
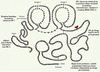 |
Figure 1. Structure et fonctions du tPA. Le tPA est une enzyme sérine-protéase pléiotrope exerçant des fonctions aussi bien au niveau vasculaire qu’au sein du parenchyme cérébral. Chacun des différents domaines du tPA, finger, growth factor, kringle 1, kringle 2 et catalytique est à l’origine d’une ou de multiples fonctions de cette sérine protéase. |
Le double jeu du tPA
Le tPA, outre son action fibrinolytique bénéfique dans le traitement des accidents vasculaires cérébraux (AVC), est aussi suspecté de contribuer aux altérations de la barrière hémato-encéphalique (BHE) et à la promotion de la toxicité neuronale. Par ailleurs, les données expérimentales montrent que le tPA vasculaire peut traverser la barrière hémato-encéphalique, atteindre le parenchyme cérébral et contribuer aux dommages post-ischémiques. Au niveau moléculaire, un modèle physiopathologique a été proposé [1] : en réponse à l’insulte ischémique, l’activité tPA augmenterait dans l’interface entre la membrane basale, les cellules endothéliales et les astrocytes périvasculaires. Le tPA, en interagissant avec les récepteurs LRP (LDL récepteurs- light density lipoproteins receptors) pourrait contribuer à l’œdème cérébral et à l’évolution vers un processus hémorragique. Ainsi, toute stratégie visant à bloquer ou diminuer l’interaction tPA/récepteur LRP aurait pour effet de limiter l’œdème cérébral, l’activation des métalloprotéinases, et par conséquent les altérations de la barrière hématoencéphalique. En 1995, alors que la FDA (Federal drug administration) était sur le point d’approuver l’utilisation du tPA dans le traitement aigu des AVC, Tsirka et ses collaborateurs rapportaient le rôle aggravant de l’activité protéolytique du tPA endogène dans la mort neuronale ischémique. En effet, le tPA est exprimé non seulement par les cellules endothéliales vasculaires [2], mais également par des cellules du cerveau, dont les neurones qui peuvent le libérer par un processus d’exocytose [3]. Cet effet délétère du tPA endogène dans le parenchyme cérébral a été confirmé par plusieurs auteurs [4, 5], suscitant le doute dans la communauté scientifique, les effets bénéfiques de la thrombolyse pouvant être limités ou masqués par les conséquences délétères. Si la plupart des études basées sur un modèle thrombotique ont confirmé que l’injection intraveineuse du tPA a un effet de protection du cerveau parce qu’il facilite la reperfusion [6], d’autres études, utilisant les modèles mécaniques d’occlusion de l’artère cérébrale moyenne et ne prenant pas en compte la composante vasculaire du tPA, ont clairement montré un renforcement des dommages ischémiques. Différentes hypothèses peuvent être avancées pour expliquer l’effet proexcitotoxique/proneurotoxique du tPA. La première se fonde sur la capacité qu’a cette protéase de convertir le plasminogène en plasmine [7]. On pense que l’action protéolytique extracellulaire de la plasmine favoriserait la dégradation de la matrice extracellulaire, le substrat essentiel des neurones [8]. Le tPA module positivement la transmission glutamatergique en favorisant la signalisation dépendante des récepteurs NMDA. En effet, le tPA peut se lier à la sous-unité NR1 du récepteur NMDA et engendrer une augmentation du flux de calcium par le récepteur activé [9].
Comment limiter les effets négatifs du tPA ?
Un effort considérable a été entrepris par la communauté scientifique pour convaincre les cliniciens que les résultats obtenus chez les patients traités avec le tPA pourraient être grandement améliorés si l’on pouvait en limiter les effets délétères au sein du parenchyme cérébral. Ceci est illustré par le succès de plusieurs stratégies appliquées à des modèles animaux. Par exemple, la coadministration du tPA avec un peptide leurre du PAI-1 (l’inhibiteur de type 1 des activateurs du plasminogène) ou avec la protéine C activée semble améliorer significativement l’effet du tPA en empêchant son effet proneurotoxique [10]. En accord avec ces données, une stratégie d’immunothérapie in vivo avec des anticorps anti-NR1 cherchant à inhiber l’interaction du tPA avec NR1 réduit les dommages ischémiques chez la souris [11]. Conceptuellement, l’agent thrombolytique idéal devrait être un agent fibrinolytique efficace et « pur », exempt d’effets nocifs sur des cellules gliales ou neuronales et sur la barrière hémato-encéphalique. La structure des dérivés du tPA apporte quelques informations : les domaines structuraux minimaux requis pour l’effet proneurotoxique du tPA correspondent au reteplase2, c’est-à-dire aux domaines K2 et SP. C’est en effet le domaine kringle 2 qui est impliqué dans le processus d’interaction avec la sous-unité NR1 du récepteur NMDA. Contrairement au tPA, DSPA (ou desmoteplase)3, qui ne contient pas de domaine kringle 2, s’est avéré in vivo exempt d’activité proneurotoxique [12]. De façon intéressante, ce domaine kringle 2 du tPA est essentiel pour que celui-ci puisse activer le pro-PDGF-cc (platelet-derived growth factor). Ce dernier, en se liant à son récepteur PDGFR-α au niveau endothélial, contribue à la perte d’intégrité de la barrière hémato-encéphalique en conditions ischémiques [13]. Récemment, un modèle reconstituant une barrière hémato-encéphalique in vitro a permis de démontrer que le desmoteplase a la capacité de traverser la barrière hémato-encéphalique intacte par un mécanisme de transcytose dépendant des récepteurs LRP. Par ailleurs, la co-administration par voie intraveineuse de DSPA et de tPA contrarie les effets proneurotoxiques du tPA parce que le DSPA entre en compétition avec le tPA pour le passage de la barrière vasculaire vers le parenchyme cérébral [12]. En conséquence, une molécule dérivée du tPA dépourvu de l’activité due au domaine K2 serait, en théorie, incapable de traverser la barrière hémato-encéphalique et d’aggraver des dommages excitotoxiques tout en conservant son action fibrinolytique bénéfique. En outre, une molécule tPA dépourvue de domaine finger réduirait également l’activation microgliale. Développer de nouveaux dérivés du tPA par l’introduction de mutations ponctuelles qui confèrent des propriétés bénéfiques ou suppriment des propriétés délètères est une voie prometteuse vers ce tPA idéal : le tenecteplase, dont la demi-vie est supérieure à celle du tPA, est un exemple illustrant cette démarche.
Conflit d’intérêts
Les auteurs déclarent n’avoir aucun conflit d’intérêts concernant les données publiées dans cet article.
NXY-059 : une drogue dont le mécanisme d’action proposé est la capture des radicaux libres dont on connaît l’action délètère en période post-ischémique, par leur induction d’une neurotoxicité et d’altérations de l’intégrité de la barrière hémato-encéphalique.
Le reteplase (rPA, Rapilysin®) dérive du tPA dont il ne conserve que les domaines kringle 2 (K2) et protéasique (SP). Cette délétion est responsable d’une liaison à la fibrine cinq fois plus faible que celle du tPA natif.
DSPA : activateurs du plasminogène isolés de la salive de la chauve-souris Desmodus. Leurs structures les rapprochent du tPA.
Références
- Polavarapu R, An J, Zhang C, Yepes M. Regulated intramembrane proteolysis of the low-density lipoprotein receptor-related protein mediates ischemic cell death. Am J Pathol 2008; 172 : 1355–62. [Google Scholar]
- Angles-Cano E, Balaton A, Le Bonniec B, et al. Production of monoclonal antibodies to the high fibrin-affinity, tissue-type plasminogen activator of human plasma. Demonstration of its endothelial origin by immunolocalization. Blood 1985; 66 : 913–20. [Google Scholar]
- Gualandris A, Jones TE, Strickland S, Tsirka SE. Membrane depolarization induces calcium-dependent secretion of tissue plasminogen activator. J Neurosci 1996; 16 : 2220–5. [Google Scholar]
- Wang YF, Tsirka SE, Strickland S, et al. Tissue plasminogen activator (tPA) increases neuronal damage after focal cerebral ischemia in wild-type and tPA-deficient mice. Nat Med 1998; 4 : 228–31. [Google Scholar]
- Nicole O, Docagne F, Ali C, et al. The proteolytic activity of tissue-plasminogen activator enhances NMDA receptor-mediated signaling. Nat Med 2001; 7 : 59–64. [Google Scholar]
- Nagai N, De Mol M, Lijnen HR, et al. Role of plasminogen system components in focal cerebral ischemic infarction : a gene targeting and gene transfer study in mice. Circulation 1999; 99 : 2440–4. [Google Scholar]
- Tsirka SE, Rogove AD, Bugge TH, et al. An extracellular proteolytic cascade promotes neuronal degeneration in the mouse hippocampus. J Neurosci 1997; 17 : 543–52. [Google Scholar]
- Chen ZL, Strickland S. Neuronal death in the hippocampus is promoted by plasmin-catalyzed degradation of laminin. Cell 1997; 91 : 917–25. [Google Scholar]
- Fernandez-Monreal M, Lopez-Atalaya JP, Benchenane K, et al. Is tissue-type plasminogen activator a neuromodulator ? Mol Cell Neurosci 2004; 25 : 594–601. [Google Scholar]
- Armstead WM, Nassar T, Akkawi S, et al. Neutralizing the neurotoxic effects of exogenous and endogenous tPA. Nat Neurosci 2006; 9 : 1150–5. [Google Scholar]
- Benchenane K, Castel H, Boulouard M, et al. Anti-NR1 N-terminal-domain vaccination unmasks the crucial action of tPA on NMDA-receptor-mediated toxicity and spatial memory. J Cell Sci 2007; 120 : 578–85. [Google Scholar]
- Lopez-Atalaya JP, Roussel BD, Ali C, et al. Recombinant Desmodus rotundus salivary plasminogen activator crosses the blood-brain barrier through a low-density lipoprotein receptor-related protein-dependent mechanism without exerting neurotoxic effects. Stroke 2007; 38 : 1036–43. [Google Scholar]
- Su EJ, Fredriksson L, Geyer M, et al. Activation of PDGF-CC by tissue plasminogen activator impairs blood-brain barrier integrity during ischemic stroke. Nat Med 2008; 14 : 731–7. [Google Scholar]
© 2009 médecine/sciences - Inserm / SRMS
Liste des figures
|
Figure 1. Structure et fonctions du tPA. Le tPA est une enzyme sérine-protéase pléiotrope exerçant des fonctions aussi bien au niveau vasculaire qu’au sein du parenchyme cérébral. Chacun des différents domaines du tPA, finger, growth factor, kringle 1, kringle 2 et catalytique est à l’origine d’une ou de multiples fonctions de cette sérine protéase. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.