")
")
| Issue |
Med Sci (Paris)
Volume 25, Number 10, Octobre 2009
Accidents vasculaires cérébraux
|
|
|---|---|---|
| Page(s) | 847 - 854 | |
| Section | M/S revues : Accidents vasculaires cérébraux (2) | |
| DOI | https://doi.org/10.1051/medsci/20092510847 | |
| Published online | 15 octobre 2009 | |
Ischémie cérébrale
Les pistes thérapeutiques de demain
Cerebral ischaemia: tomorrow’s therapeutic tracks
1
EA1046, département de pharmacologie médicale, Institut de médecine prédictive et de recherche thérapeutique, Université Lille-Nord de France, Faculté de médecine, Centre hospitalier et universitaire de Lille, 2, avenue Oscar Lambret 59037 Lille Cedex, France
2
U861, I-Stem, Genopole Campus 1, 5, rue Henri Desbruères, 91030 Évry Cedex, France
3
UMR-CNRS 7102, Université Pierre et Marie Curie, Faculté des sciences Jussieu, Paris, France
4
UMR 6097, Institut de pharmacologie moléculaire et cellulaire, CNRS, Université Nice-Sophia Antipolis, Valbonne, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Les stratégies thérapeutiques utilisées au décours des accidents vasculaires cérébraux (AVC) reposent actuellement sur la thrombolyse, utilisée dans les toutes premières heures afin d’assurer la reperfusion du tissu cérébral ischémié. Des approches de protection cérébrale seraient cependant nécessaires avec un double objectif : servir de traitement adjuvant à la thrombolyse cérébrale pour en allonger la fenêtre thérapeutique ; limiter l’extension des lésions ischémiques en modulant les processus physiopathologiques. Si de nombreuses pistes se sont révélées infructueuses, la recherche d’agents protecteurs cérébraux se poursuit en privilégiant le développement de molécules aux effets pléiotropes ciblant l’ensemble des acteurs de l’unité neurovasculaire tant les mécanismes moléculaires et cellulaires sont intriqués au cours de l’ischémie cérébrale. Des espoirs naissent également de l’observation d’une récupération fonctionnelle et d’une réparation spontanées qui pourraient être amplifiées par des approches pharmacologiques ou de thérapie cellulaire.
Abstract
Thrombolysis remains the main therapeutic strategy used in stroke, but with a limited use to only a part of stroke patients. A neuroprotective approach would be necessary with a double objective : (1) to serve as an add-on treatment with thrombolysis to improve safety and increase therapeutic window ; (2) to limit infarct area by delaying neuronal death. While numerous molecules failed in clinical trials in stroke, pharmacological development is ongoing with pleiotropic drugs targeting both neuronal and vascular parts of neurovascular unit. Another approach targets the functional rehabilitation and the neurorepair using pharmacological ways or cell therapy.
© 2009 médecine/sciences - Inserm / SRMS
Compte tenu de son incidence et de sa sévérité, la pathologie vasculaire cérébrale demeure un enjeu thérapeutique majeur. Des progrès indéniables ont été faits dont témoigne une diminution progressive, au cours des deux dernières décennies, de l’incidence des AVC. C’est le résultat de la validation de traitements préventifs dont l’efficacité a été établie par de larges études de morbi-mortalité au niveau de preuve important [1]. Au-delà d’un effet sur l’incidence, certains traitements préventifs exercent un effet de neuroprotection dont témoigne une moindre sévérité de l’accident vasculaire lorsqu’il survient chez les patients traités. Ce double effet, sur l’incidence et la neuroprotection, justifie un développement thérapeutique spécifique plus important eu égard au nombre de patients à risque ; celui-ci doit s’appuyer au plan pharmacologique sur les mécanismes de protection endogène mis en évidence par les expériences de préconditionnement.
Les limites de la thombolyse
Cependant, si le traitement préventif est efficace, il ne prévient qu’une partie des accidents ischémiques ; le développement parallèle de stratégies thérapeutiques, soit à la phase aiguë soit plus tard pour accélérer la récupération fonctionnelle, doit être une priorité (Figure 1). Le seul traitement dont l’utilisation est validée est la fibrinolyse, applicable dans les quatre heures trente qui suivent le début de l’accident ischémique. Elle assure une reperfusion rapide du tissu ischémié permettant d’accroître le nombre de patients vivants et autonomes [2]. Sa limite réside dans cette fenêtre thérapeutique étroite, car au-delà de ce délai recommandé de 4h30, le risque hémorragique est élevé. La proportion de patients susceptibles de bénéficier d’une thrombolyse est de ce fait inférieure à 10 %. Cette approche nécessite le développement de nouveaux agents fibrinolytiques ou le recours à des traitements adjuvants permettant de limiter le risque hémorragique d’une fibrinolyse administrée au-delà du délai recommandé [2].
 |
Figure 1. Cinétique des mécanismes physiopathologiques de l’ischémie et principales approches pharmacologiques (d’après [15]). |
Les stratégies de neuroprotection
Ces traitements adjuvants pourraient aussi être des agents neuroprotecteurs, c’est-à-dire des agents capables de retarder ou de prévenir la mort neuronale : leur recherche a débuté dans les années 1980 mais reste aujourd’hui infructueuse en dépit du nombre de molécules testées tant au plan expérimental que clinique depuis une vingtaine d’années [3]. Ces échecs répétés conduisent aujourd’hui à développer des stratégies pharmacologiques aux mécanismes pléiotropes et ciblant l’ensemble de l’unité neurovasculaire. Plusieurs revues de synthèse s’accordent sur le fait que la prochaine étape dans la compréhension de la physiopathologie de l’ischémie cérébrale nécessite une vision intégrée de l’ensemble des mécanismes à l’échelon de l’unité neurovasculaire, en mettant en lumière les relations mécanistiques entre les trois compartiments cellulaires : l’endothélium, les astrocytes, et les neurones [4].
De nouvelles approches pourraient tirer parti de l’observation de la mise en jeu de mécanismes protecteurs endogènes dont l’effet est complété par la mobilisation des cellules souches d’origine cérébrale ou hématopoïétique, qui permet une migration et une différenciation de nouvelles cellules dans le foyer ischémique [5, 6]. Ces mécanismes endogènes, bénéfiques parce qu’ils limitent les conséquences de l’ischémie, requièrent également une coopération entre les différents compartiments de l’unité neurovasculaire. Les travaux menés sur les mécanismes protecteurs endogènes et sur les phénomènes de plasticité cérébrale conduisent à tester de nouvelles approches favorisant la récupération fonctionnelle ou la réparation cérébrale post-ischémique ; c’est le cas des essais de thérapie cellulaire. Plusieurs équipes de recherche françaises contribuent à faire émerger ces nouvelles cibles pharmacologiques et ces nouvelles approches thérapeutiques [7].
Du préconditionnement à la neuroprotection préventive
Le concept de préconditionnement recouvre l’ensemble des stratégies expérimentales qui consistent à induire un phénotype protecteur avant que le stress ischémique ne survienne [8]. Une preuve de la pertinence clinique du préconditionnement cérébral a été apportée par la démonstration que chez les patients ayant présenté un accident ischémique transitoire - situation clinique mimant un préconditionnement ischémique - la sévérité des accidents ischémiques constitués survenant au décours et même très à distance de l’accident ischémique transitoire était moindre [5]. La programmation de la résistance à l’ischémie vise à : (1) activer des voies moléculaires qui confèrent des mécanismes de défenses endogènes vis-à-vis de l’agression ischémique ; (2) prévenir, par la répression de l’activité des gènes correspondants impliqués, l’activation des voies moléculaires qui jouent, au cours de l’ischémie, un rôle essentiel dans l’induction de la mort neuronale nécrotique ou apoptotique [9]. De multiples effecteurs sont mis en jeu dans ce processus : des protéines de structure ou de fonction de la membrane cellulaire, du cytosquelette, de la mitochondrie ou des organites impliqués dans le métabolisme cellulaire [8]. On peut distinguer deux types d’effecteurs : (1) ceux qui sont le résultat d’une programmation génomique des fonctions de survie : protéines anti-apoptotiques (Bcl-2, UCP-2, IAP inhibitors of apoptosis proteins), enzymes de réparation de l’ADN, gènes de survie, enzymes anti-oxydantes, facteurs neurotrophiques et de croissance, érythropoïétine, protéines chaperonnes (HSP pour heat shock protein etc.) ; (2) ceux qui résultent d’une programmation génomique de l’inhibition des phénomènes de mort cellulaire : protéines pro-apoptotiques (Bax, cytochrome c et AIF), facteurs de transcription pro-inflammatoires (NFκB), enzymes de production de radicaux libres, endonucléases, p53, c-jun, etc. Si longtemps le préconditionnement a été considéré comme un mécanisme essentiellement neuronal, il est maintenant reconnu comme un phénomène de protection globale des trois compartiments de l’unité neurovasculaire [10]. Les mécanismes précédemment cités ont été souvent décrits à l’échelon neuronal et le préconditionnement induit également une augmentation de la neurogenèse. Mais il est également capable de préserver la fonction endothéliale avec pour conséquence un maintien du débit sanguin cérébral et des capacités d’angiogenèse. Les astrocytes et les cellules microgliales sont également transitoirement activés par le préconditionnement.
Dans la continuité des travaux sur le préconditionnement, l’effet neuroprotecteur préventif de classes médicamenteuses utilisées dans la prévention primaire ou secondaire des accidents ischémiques cérébraux a été testé. Une série de travaux a permis de montrer que l’administration préventive de différentes statines était associée à une diminution de la taille des infarctus dans des modèles expérimentaux, à une augmentation de l’expression de la NOS (nitric oxide synthase) endothéliale et à un effet anti-oxydant [11]. Les antagonistes des récepteurs AT1 de l’angiotensine 2 (sartans) ont également des effets similaires, par leur modulation des voies impliquées dans le phénomène de préconditionnement cérébral. Un effet identique a été également décrit avec la classe des fibrates [12]. Hormis les médicaments, l’exercice physique ou la prise d’oméga-3 induisent expérimentalement un effet neuroprotecteur préventif [13]. Des résultats expérimentaux complémentaires ont démontré que l’effet de ces différentes classes, en particulier celle des statines, ne persistait pas au-delà de quelques jours après l’arrêt de l’administration de la molécule, avant la réalisation de l’ischémie, suggérant que l’effet neuroprotecteur préventif n’est pas lié à un mécanisme de préconditionnement mais plus à la présence du médicament dans l’organisme au moment de la survenue de l’accident ischémique cérébral. Il n’en demeure pas moins que, quel que soit le mécanisme en jeu, la neuroprotection préventive reste cliniquement pertinente puisque plusieurs études cliniques ont permis de montrer que les patients traités par des agents hypolipémiants ou pratiquant un exercice physique régulier avant la survenue d’un accident ischémique cérébral ont une sévérité moindre des symptômes cliniques [14].
De nouvelles cibles pour une neuroprotection à la phase aiguë ?
Même si le concept de neuroprotection vis-à-vis des lésions ischémiques est né au cours des années 1980 avec les premières recherches sur la physiopathologie de l’ischémie cérébrale, ce n’est que dix ans plus tard que la recherche sur la neuroprotection a vraiment émergé, comme l’atteste l’afflux de publications référencées. Au cours des six dernières années, 1 000 articles expérimentaux et 400 articles cliniques ont été publiés dans ce domaine [3]. La neuroprotection peut être définie comme une stratégie ou une combinaison de stratégies qui peuvent inhiber, interrompre ou ralentir les événements moléculaires et cellulaires qui conduisent à des lésions ischémiques irréversibles ; elle exclue les thérapeutiques visant à agir directement sur l’obstruction artérielle elle-même. S’appuyant sur des modèles expérimentaux reproduisant la pathologie vasculaire cérébrale et sur les nombreux mécanismes physiopathologiques mis en évidence (excitotoxicité, dépolarisation péri-infarctus, stress oxydant, inflammation, activation des caspases…), de nombreux agents neuroprotecteurs ont été évalués dans des modèles précliniques mais aussi en clinique [15]. Les échecs répétés de ces essais ont conduit à remettre en question la pertinence de la stratégie, des modèles précliniques ou de l’évaluation clinique (pour une discussion de ces points voir [16]). Si les recommandations STAIR (Stroke therapy academic industry roundtable)1 [17] pour l’évaluation préclinique ont permis d’améliorer certaines étapes du développement de molécules thérapeutiques, un effort doit être encore fait sur les étapes translationnelles [18], avec notamment la prise en compte de l’évolution cinétique et spatiale de la cible testée, afin de mieux sélectionner les patients (Tableau I). Le développement du NXY059, un agent anti-oxydant dont le développement pré-clinique s’est fait selon les critères de recommandation STAIR, est un bon exemple des efforts qui sont encore nécessaires pour réussir à obtenir des produits efficaces chez l’homme. La possibilité de tester précocement des combinaisons de traitements serait nécessaire, même si elle pose des problèmes au plan industriel. Bien que cette stratégie de neuroprotection se soit heurtée à de nombreuses déconvenues, un certain nombre de pistes pharmacologiques restent exploitées ; elles proposent des approches innovantes pour cibler les principales voies physiopathologiques, en particulier celles qui interviennent de manière différée par rapport au début de l’ischémie (Tableau II).
Recommandations STAIR et modifications proposées lors de la dernière table ronde. PA : pression artérielle ; IRM : imagerie par résonance magnétique (d’après [17]).
Principales molécules dont le développement dans les accidents vasculaires cérébraux a été arrêté ou est en cours.
Trois pistes pour l’avenir
Antagoniser l’excitotoxicité
La voie de l’excitotoxicité liée à la libération massive de glutamate reste une cible intéressante dans la mesure où elle est responsable de lésions sévères et rapides et où le tPA, qui clive le récepteur du glutamate NMDA (N-methyl d-aspartate receptor) aggrave ses conséquences [36] (→). Bien que certains antagonistes des récepteurs ionotropiques du glutamate soient encore en phase de développement, cette approche s’est révélée être un échec, et les recherches s’orientent plutôt vers une approche fondée sur une immunothérapie. L’administration d’anticorps dirigés contre certaines régions du récepteur NMDA est actuellement à l’étude avec des résultats encourageants : une prévention des lésions par des anticorps ciblant le clivage du récepteur NMDA par le tPA a été observée avec une diminution des conséquences délétères [19].
(→) Voir l’article D. Vivien, p. 855 de ce numéro
Antagoniser la dépolarisation neuronale
Les mécanismes de la mort neuronale impliquent aussi la dépolarisation neuronale, en partie expliquée par des modifications des canaux sodiques ou calciques voltage-dépendants ; la modulation pharmacologique de ces canaux n’est toutefois pas efficace au plan thérapeutique, en dépit de résultats précliniques positifs. La piste d’une intervention ciblant les canaux ioniques, en particulier les différentes sous-familles de canaux potassiques décrites ces dernières années, reste toutefois intéressante à exploiter. Parmi les canaux potassiques étudiés, les canaux potassiques rectifiant-entrants (Kir) ou voltage-dépendants (Kv) pourraient être des cibles à moduler indirectement via le stress oxydant ou certaines voies de transduction intracellulaires [10]. Mais les canaux de la famille TREK (TWIK1-related K+ channel)/TRAAK (TWIK related arachidonic acid activated K+ channel) se révèlent être les plus prometteurs dans la mesure où ils régulent des fonctions neuronales, astrocytaires et endothéliales et qu’ils sont modulés par le riluzole2 et les oméga-3 contribuant aux effets neuroprotecteurs de ces deux types d’agents [20]. Les phénomènes inflammatoires et oxydatifs semblent pouvoir être modulés directement mais aussi indirectement via des facteurs de transcription comme NFκB ou la PARP (Poly ADP-ribose polymerase) [21].
Inhiber l’apoptose
L’observation que, dans la zone de pénombre ischémique, les neurones meurent par apoptose a stimulé la recherche d’inhibiteurs d’apoptose, même si les résultats obtenus avec cette stratégie restent aujourd’hui discutés [22]. L’apoptose est probablement un processus trop complexe pour constituer en tant que tel une cible pharmacologique ; les recherches récentes s’orientent plus vers la compréhension du rôle des caspases dans la mort neuronale survenant de manière retardée et la recherche d’inhibiteurs ciblés, ou le renforcement d’inhibiteurs endogènes [23]. Cette dernière stratégie a été particulièrement développée avec l’un des plus puissants inhibiteurs d’apoptose endogènes, le XIAP (X-linked inhibitor of apoptosis protein), dont la vectorisation, favorisant l’accumulation cérébrale, est associée à un effet protecteur cérébral [24, 25].
Vers des agents pléiotropes ciblant l’unité neurovasculaire ?
Les échecs rencontrés dans le développement des agents neuroprotecteurs ont conduit à envisager des stratégies ciblant les mécanismes physiopathologiques de façon plus intégrée ou utilisant des agents pharmacologiques à l’action pléiotrope. La plupart des recherches thérapeutiques dans les AVC se sont concentrées initialement sur le compartiment neuronal, alors que l’ischémie cérébrale touche l’intégralité de l’unité neurovasculaire qui intègre l’endothélium, les astrocytes, et les neurones (Figure 2). Chacun de ces acteurs de l’unité neurovasculaire est affecté par l’activation des différentes voies physiopathologiques déclenchées par l’ischémie cérébrale ; les interactions cellulaires qui existent au sein de l’unité neurovasculaire expliquent pourquoi les altérations post-ischémiques de l’un des compartiments cellulaires ont des répercussions sur les autres compartiments [4, 10, 26].
Les cellules gliales et microgliales
Les astrocytes sont des déterminants critiques dans la physiopathologie de l’ischémie cérébrale puisqu’ils assurent le couplage entre les neurones et les vaisseaux [27]. Ils peuvent assurer une protection à court terme en libérant des facteurs de croissance ou à plus long terme par la libération d’autres molécules qui facilitent la neurogenèse et la régénération. Cependant la sévérité de l’ischémie peut compromettre les fonctions astrocytaires et la survie neuronale post-ischémique, en raison d’une diminution de l’apport d’énergie aux neurones et d’une contribution à la propagation de la dépolarisation péri-infarctus. Les cellules microgliales sont également activées au cours du processus d’ischémie cérébrale et exercent des effets contrastés voire contradictoires : (1) un effet délétère a été décrit, par le biais d’une libération de substances toxiques pour les neurones (cytokines, radicaux libres, médiateurs inflammatoires…), expliquant l’effet neuroprotecteur de la minocycline ou de l’édaravone qui inhibent l’activation microgliale ; (2) des données plus récentes suggèrent que les cellules microgliales pourraient exercer un effet neuroprotecteur [28].
Le compartiment vasculaire
Le compartiment vasculaire a été longtemps négligé alors que la réponse des neurones et des microvaisseaux adjacents au processus d’ischémie est probablement simultanée et coordonnée compte tenu des liens physiques étroits qui existent au sein de l’unité neurovasculaire [4]. Très rapidement au décours du processus d’ischémie-reperfusion, les vaisseaux cérébraux répondent de façon dynamique par une ouverture de la barrière hémato-encéphalique, l’expression de marqueurs d’adhésion des leucocytes, l’effondrement de l’unité fonctionnelle neuro-glio-vasculaire, une diminution de la réactivité endothélium-dépendante et la dégradation de la matrice extracellulaire. Toutes ces altérations influençent la viabilité des cellules avoisinantes et l’intégrité fonctionnelle des relations entre les apports vasculaires et l’activité neuronale et gliale [4]. Le stress oxydant joue un rôle important par ses effets toxiques sur les constituants de l’unité neurovasculaire mais aussi en raison d’une diminution de la biodisponibilité du NO (Figure 3). Ce rôle explique l’intérêt du développement d’agents anti-oxydants, comme le NXY059, pour leur double effet de protecteur neuronal et vasculaire. Mais les phénomènes inflammatoires semblent jouer le rôle prépondérant, via les polynucléaires neutrophiles ; ces cellules, à la fois circulantes et infiltrantes, sont des médiateurs des interactions neurovasculaires [29]. La déplétion en polynucléaires neutrophiles (vinblastine, anticorps anti-neutrophiles), le ciblage de facteurs attractants (MCP-1, monocyte chemotactic protein 1) ou l’inhibition des protéines d’adhésion permettent d’induire une double protection neuronale et vasculaire [30]. Les mécanismes moléculaires à l’origine de l’effet délétère des polynucléaires neutrophiles font intervenir les protéases, en particulier MMP-2 et MMP-9, qui participent aux complications hémorragiques de la thrombolyse, soulignant l’intérêt potentiel de la modulation des polynucléaires neutrophiles comme traitement adjuvant à la phase aiguë [31].
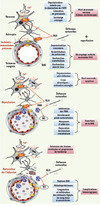 |
Figure 3. Séquence des mécanismes inflammatoires et oxydatifs au sein de l’unité neurovasculaire après un AVC. BHE : barrière hémato-encéphalique ; RLO : radicaux libres oxygénés ; PNN : polynucléaire neutrophile. |
Intérêt des molécules exerçant une action pléiotrope
Ces interactions au sein de l’unité neurovasculaire et les multiples voies physiopathologiques à l’œuvre dans les lésions ischémiques expliquent la nécessité de développer des agents pharmacologiques pléiotropes, c’est-à-dire aux effets cellulaires et moléculaires multiples. Des molécules déjà utilisées en thérapeutique humaine pour plusieurs mécanismes d’action ont été testées dans des modèles pré-cliniques d’AVC et ont montré un effet protecteur cérébral : des anti-hypertenseurs, des hypolipémiants, les thiazolidinediones, la citicoline (un dérivé de la cytidine), le G-CSF (granulocyte colony-stimulating factor), l’albumine [32]. Parmi ces classes médicamenteuses, les agents hypolipémiants, certains antihypertenseurs (les antagonistes des récepteurs de l’angiotensine 2) ou les thiazolidinediones agissent sur un sous-type de récepteurs nucléaires, les récepteurs PPAR (peroxisome proliferator-activated receptors), qui sous-tend le caractère pléiotrope de l’action de ces agents pharmacologiques. Les trois isoformes des récepteurs PPAR (PPAR-α, PPAR-β/δ et PPAR-γ) sont exprimées par les trois compartiments de l’unité neurovasculaire. Ces récepteurs nucléaires ont d’abord été caractérisés pour leurs effets sur la régulation du métabolisme : métabolisme lipidique pour les PPAR-α qui sont la cible des fibrates, des agents hypolypémiants ; métabolisme glucidique pour les PPAR-γ qui sont la cible des thiazolidinediones (ou glitazones), antidiabétiques oraux. Au-delà de leurs effets métaboliques, les récepteurs PPAR régulent les processus inflammatoires et oxydatifs par leur action sur les facteurs de transcription, comme NFκB. Ils ont montré des effets neuroprotecteurs à la phase aiguë d’AVC créés dans des modèles précliniques, effets attribués à des mécanismes anti-inflammatoires et anti-oxydants s’exerçant dans les trois compartiments neuronal, glial et vasculaire [11].
Approches thérapeutiques au décours de l’accident vasculaire :effet sur la récupération fonctionnelle et/ou la réparation
Contrairement à une idée reçue, dans la pathologie ischémique cérébrale, tout n’est pas joué dans les premières heures, même si la rapidité de l’intervention est indispensable pour préserver le maximum de tissu cérébral. L’observation clinique suggère que dans les jours et semaines qui suivent la survenue d’un accident vasculaire cérébral, une récupération fonctionnelle est encore possible, par exemple sous l’effet de la kinésithérapie. Cette capacité de récupération est probablement liée à la plasticité cérébrale, favorisée par la mise en jeu spontanée de mécanismes endogènes, dont celle des facteurs neurotrophiques comme le BDNF (brain-derived neurotrophic factor) qui améliore la récupération fonctionnelle [33]. Compte tenu des difficultés d’utilisation thérapeutique des facteurs neurotrophiques (passage de la barrière hémato-encéphalique, risques inhérents à une administration prolongée de facteurs de croissance), les recherches se sont portées sur l’évaluation de médicaments agissant directement ou indirectement sur ces facteurs neurotrophiques, comme les antidépresseurs inhibiteurs de la recapture de la sérotonine ou le méthylphénidate dont l’effet sur la récupération fonctionnelle au décours des accidents vasculaires cérébraux ischémiques est en cours d’évaluation [34]. Au décours d’une ischémie cérébrale, une mobilisation spontanée de cellules souches d’origine neurale ou issues de la moelle osseuse a été décrite, qui permet une réparation partielle du tissu cérébral lésé, puisque ces cellules souches sont capables de migrer dans le foyer ischémique et de s’y différencier en neurones, astrocytes ou cellules endothéliales. On pourrait donc imaginer amplifier cette réparation spontanée qui est insuffisante pour assurer une récupération significative [6]. Deux approches ont été proposées : l’une utilise la thérapie cellulaire par administration de cellules souches par voie systémique. Des travaux précliniques ont été concluants et des études cliniques sont en cours qui visent à mieux définir les procédures (type de cellule, stade de différenciation, quantité, voie d’administration…) et à en évaluer l’efficacité [35]. L’autre approche est pharmacologique et son objectif est de mobiliser les cellules souches endogènes vers le foyer ischémique dans une perspective de réparation du tissu cérébral. Cette approche en est encore au stade de la recherche fondamentale, essayant de comprendre les mécanismes en jeu dans les phénomènes spontanés. Certains agents pharmacologiques ayant des effets protecteurs cérébraux (G-CSF, statines…) agissent sur les cellules souches, ce qui pourrait en partie expliquer leurs effets bénéfiques [32].
Conflit d’intérêts
Les auteurs déclarent n’avoir aucun conflit d’intérêts concernant les données publiées dans cet article.
Le groupe STAIR (créé en 1999, http://thestair.org) inclut des chercheurs renommés du domaine appartenant aussi bien au secteur académique qu’à l’industrie ; ils se réunissent pour des conférences de consensus (six depuis 1999) dont l’objectif est de faire progresser les approches thérapeutiques dans le domaine des accidents vasculaires cérébraux.
Le riluzole bloque les canaux sodiques dépendants du voltage et diminue ainsi la libération présynaptique du glutamate ; cette molécule exerce un effet positif sur l’évolution de la sclérose latérale amyotrophique.
Références
- Donnan GA, Fisher M, Macleod M, Davis SM. Stroke. Lancet 2008; 371 : 1612–32. [Google Scholar]
- Hacke W, Kaste M, Bluhmki E, et al. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med 2008; 359 : 1317–29. [Google Scholar]
- Ginsberg MD. Neuroprotection for ischemic stroke: past, present and future. Neuropharmacology 2008; 55 : 363–89. [Google Scholar]
- Del Zoppo GJ. Inflammation and neurovascular unit in the setting of focal cerebral ischemia. Neuroscience 2009; 158 : 972–82. [Google Scholar]
- Shpargel KB, Jalabi W, Jin Y, et al. Preconditioning paradigms and pathways in the brain. Cleve Clin J Med 2008; 75 : S77–82. [Google Scholar]
- Hess DC, Borlongan CV. Cell-based therapy in ischemic stroke. Expert Rev Neurother 2008; 8 : 1193–201. [Google Scholar]
- Endres M, Engelhardt B, Koistinaho J, et al. Improving outcome after stroke : overcoming the translational roadblock. Cerebrovasc Dis 2008; 25 : 268–78. [Google Scholar]
- Obrenovitch TP. Molecular physiology of preconditioning-induced brain tolerance to ischemia. Physiol Rev 2008; 88 : 211–47. [Google Scholar]
- Stenzel-Poore MP, Stevens SL, King JS, Simon RP. Preconditioning reprograms to response to ischemic injury and primes the emergence of unique endogenous neuroprotective phenotypes : a speculative synthesis. Stroke 2007; 38 (suppl 2) : 680–5. [Google Scholar]
- Bastide M, Ouk T, Plaisier F, et al. Neurogliovascular unit after cerebral ischemia: is the vascular wall a pharmacological target. Psychoneuroendocrinology 2007; 32 : S36–9. [Google Scholar]
- Goldstein LB. Statins and ischemic stroke severity: cytoprotection. Curr Atheroscler Rep 2009; 11 : 296–300. [Google Scholar]
- Bordet R, Ouk T, Petrault O, et al. PPAR: a new pharmacological target for neuroprotection in stroke and neurodegenerative diseases. Biochem Soc Trans 2006; 34 : 1341–6. [Google Scholar]
- Blondeau N, Widmann C, Lazdunski M, Heurtaux C. Polyunsaturated fatty acids induce ischemic and epileptic tolerance. Neuroscience 2002; 109 : 231–41. [Google Scholar]
- Deplanque D, Masse I, Lefebvre C, et al. Prior TIA, lipid-lowering drug use, and physical activity decrease ischemic stroke severity. Neurology 2006; 67 : 1403–10. [Google Scholar]
- Zaleska MM, Mercado ML, Chavez J, et al. The development of stroke therapeutics: promising mechanisms and translational challenges. Neuropharmacology 2009; 56 : 329–41. [Google Scholar]
- Bordet R, Lestage P, Onteniente B. The concept of neuroprotective agents as a treatment modulators in the development of brain diseases. Therapie 2007; 62 : 463–72. [Google Scholar]
- Fisher M, Feuerstein G, Howells DW, et al. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke 2009; 40 : 2244–50. [Google Scholar]
- Feuerstein GZ, Zaleska MM, Krams M, et al. Missing steps in the STAIR case : a translational medicine perspective on the development of NXY-059 for treatment of acute ischemic stroke. J Cereb Blood Flow Metab 2008; 28 : 217–9. [Google Scholar]
- Benchenane K, Castel H, Boulouard M, et al. Anti-NR1 N-terminal-domain vaccination unmasks the crucial action of tPA on NMDA-receptor-mediated toxicity and spatial memory. J Cell Sci 2007; 120 : 578–85. [Google Scholar]
- Heurtaux C, Laigle C, Blondeau N, et al. Alpha-linolenic acid and riluzole treatment confer cerebral protection and improve survival after focal brain ischemia. Neuroscience 2006; 137 : 241–51. [Google Scholar]
- Haddad M, Rhinn H, Bloquel C, et al. Anti-inflammatory effects of PJ34, a poly (ADP-ribose) polymerase inhibitor, in transcient focal cerebral ischemial in mice. Br J Pharmacol 2006; 149 : 23–30. [Google Scholar]
- Chauvier D, Ankri S, Charriaut-Marlangue C, et al. Broad-spectrum caspase inhibitors : from myth to reality ? Cell Death Differ 2007; 14 : 387–91. [Google Scholar]
- Onténiente B. Natural and synthetic inhibitors of caspases : targets for novel drugs. Curr Drug Targets CNS Neurol Disord 2004; 3 : 333–40. [Google Scholar]
- Guégan C, Braudeau J, Couriaud C, et al. PTD-XIAP protects against cerebral ischemia by anti-apoptotic and transcriptional regulatory mechanisms. Neurobiol Dis 2006; 22 : 177–86. [Google Scholar]
- Spahn A, Blondeau N, Heurteaux C, et al. Concomitant transitory up-regulation of X-linked inhibitor of apoptosis protein (XIAP) and the heterogeneous nuclear ribonucleoprotein C1-C2 in surviving cells during neuronal apoptosis. Neurochem Res 2008; 33 : 1859–68. [Google Scholar]
- Lok J, Gupta P, Guo S, et al. Cell-cell signalling in neurovascular unit. Neurochem Res 2007; 32 : 2032–45 [Google Scholar]
- Villapol S, Gelot A, Renolleau S, Charriaut-Marlangue C. Astrocyte responses after neonatal ischemia : the yin and the yang. Neuroscientist 2008; 14 : 339–44. [Google Scholar]
- Amantea D, Nappi G, Bernardi G, et al. Post-ischemic brain damage : pathophysiology and role of inflammatory mediators. FEBS J 2009; 276 : 13–26. [Google Scholar]
- Fisher M. Injuries to the vascular endothelium: vascular wall and endothelial dysfunction. Rev Neurol Dis 2008; 5 : S4–11. [Google Scholar]
- Gautier S, Ouk T, Petrault O, et al. Neutrophils contribute to intracerebral haemorrhages after treatment with recombinant tissue plasminogen activator following cerebral ischaemia. Br J Pharmacol 2009; 156 : 673–9. [Google Scholar]
- Zhao BQ, Tejima E, Lo EH. Neurovascular proteases in brain injury, hemorrhage and remodeling after stroke. Stroke 2007; 38 (suppl 2) : 748–52. [Google Scholar]
- Minnerup J, Schäbitz WR. Multifunctional actions of approved and candidate stroke drugs. Neurotherapeutics 2009; 6 : 43–52. [Google Scholar]
- Rodriguez-Gonzalez R, Hurtado O, Sobrino T, Castillo J. Neuroplasticity and cellular therapy in cerebral infarction. Cerebrovasc Dis 2007; 24 : S167–80. [Google Scholar]
- Tardy J, Pariente J, Leger A, et al. Methylphenidate modulates cerebral post-stroke reorganization. Neuroimage 2006; 33 : 913–22. [Google Scholar]
- Bacigaluppi M, Pluchino S, Martino G, et al. Neural stem/precursor cells for the treatment of ischemic stroke. J Neurol Sci 2008; 265 : 73–7. [Google Scholar]
- Vivien D, Gauberti M, Guedin P, Anglés-Cano E. Comment optimiser l’utilisation du tPA ? Med Sci (Paris) 2009; 25 : 855–7. [Google Scholar]
Liste des tableaux
Recommandations STAIR et modifications proposées lors de la dernière table ronde. PA : pression artérielle ; IRM : imagerie par résonance magnétique (d’après [17]).
Principales molécules dont le développement dans les accidents vasculaires cérébraux a été arrêté ou est en cours.
Liste des figures
|
Figure 1. Cinétique des mécanismes physiopathologiques de l’ischémie et principales approches pharmacologiques (d’après [15]). |
| Dans le texte | |
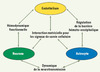 |
Figure 2. Schéma des interactions au sein de l’unité neurovasculaire (d’après [26]). |
| Dans le texte | |
|
Figure 3. Séquence des mécanismes inflammatoires et oxydatifs au sein de l’unité neurovasculaire après un AVC. BHE : barrière hémato-encéphalique ; RLO : radicaux libres oxygénés ; PNN : polynucléaire neutrophile. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.