")
")
| Issue |
Med Sci (Paris)
Volume 25, Number 6-7, Juin-Juillet 2009
|
|
|---|---|---|
| Page(s) | 555 - 557 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/2009256-7555 | |
| Published online | 15 juin 2009 | |
Du nouveau sur la régulation de la réparation de l’ADN par excision de nucléotides
New insight into the regulation of DNA nucleotide excision repair: implications for cancer development and treatment
Faculté de médecine, Université de Montréal et Centre de recherche, Hôpital Maisonneuve-Rosemont, 5145, boulevard de l’Assomption, Montréal, Québec, H1T 2M4 Canada
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
La réparation par excision de nucléotides (nucleotide excision repair, NER) corrige une grande variété de lésions de l’ADN qui provoquent une distorsion de la double hélice et qui, par conséquent, bloquent la transcription et la réplication [1] (voir Figure 1 pour une description détaillée). Ces lésions peuvent être causées par une multitude d’agents environnementaux. Entre autres, les CPD (dimères de cyclobutane-pyrimidine) et les 6-4PP (pyrimidine (6-4) pyrimidone), provoqués par les rayons ultraviolets solaires (UV), sont les principaux facteurs étiologiques du cancer de la peau [2]. La maladie récessive autosomique Xeroderma pigmentosum (XP) illustre de façon exemplaire l’importance de la NER. Cette perturbation génétique est caractérisée par un défaut de la NER engendré par des mutations affectant des gènes (XP-A à XP-G) directement impliqués dans cette voie de réparation. Les personnes atteintes manifestent une hypersensibilité aux UV (coups de soleil sévères, pigmentation anormale, troubles oculaires) ainsi qu’une très forte prédisposition au développement de tumeurs cutanées. L’âge médian de survenue du premier cancer est d’environ 8 ans. De plus, près du tiers des patients présente des affections neurologiques sévères comme une microcéphalie et des troubles du développement [3].
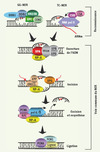 |
Figure 1. Réparation par excision de nucléotides. La NER procède selon deux sous-voies chevauchantes qui se distinguent uniquement à l’étape de reconnaissance de la lésion : (1) la réparation globale (global genomic NER, GG-NER) qui éradique des lésions dans l’ensemble du génome et (2) la réparation couplée à la transcription (transcription coupled NER ; TC-NER) qui agit uniquement sur le brin transcrit des gènes actifs. Lors de la GG-NER, via leurs capacités à déceler d’importantes déformations structurales, l’hétérodimère DDB1-DDB2 et/ou le complexe hétérotrimérique XPC-HR23B-CEN2 détectent les lésions. Alternativement, lors de la TC-NER, le blocage de l’ARN polymérase au site endommagé émet le signal à l’origine du recrutement des protéines CSA (Cockayne syndrome A protein) et CSB. À la suite de l’une ou l’autre de ces étapes de reconnaissance, la voie commune de la NER est mobilisée séquentiellement de la manière suivante : (1) les ADN hélicases (XPB et XPD), composantes du facteur de transcription TFIIH, déroulent la double hélice d’ADN ; (2) les protéines RPA (replication protein A) et XPA sont sollicitées afin de permettre la stabilisation du complexe et le recrutement des facteurs subséquents ; (3) l’arrivée des endonucléases XPG et XPF-ERCC1 mène à l’incision de la lésion en 3’ et 5’ respectivement et l’oligonucléotide endommagé (~20 bp) est excisé ; (4) l’ADN est resynthétisé par des facteurs de réplication, incluant les polymérases ε/δ, et finalement lié à nouveau par des ligases. |
Depuis sa découverte, la NER a été très étudiée et le processus reconstitué in vitro. Néanmoins, sa régulation demeure mal comprise. En réponse aux UV (et aux autres agents provoquant un stress réplicatif), la kinase ATR (Ataxia telangiectasia and rad 3-related) est activée et joue un rôle protecteur considérable [4]. Entre autres, ATR phosphoryle le suppresseur de tumeur p53 [5], un important régulateur du cycle cellulaire et de l’apoptose, également requis pour une NER efficace [6]. Mis à part p53, ATR phosphoryle une pléthore de protéines intervenant de façon critique dans la réponse aux stress génotoxiques et dont certaines sont directement associées à la NER (XP-A, RP-A) [7]. Toutefois, aucune étude n’avait jusqu’à maintenant montré l’efficacité de la NER dans des cellules humaines dont l’activité d’ATR est inhibée. Par conséquent, nous avons émis l’hypothèse qu’ATR puisse participer à la NER, et ce spécifiquement au cours de la phase S du cycle cellulaire conformément à son rôle prééminent durant la réplication de l’ADN.
Cependant, en raison de diverses considérations techniques, d’une façon générale, les méthodes traditionnelles d’étude de la NER excluent les cellules en phase S de l’analyse. Pour pallier ce problème, nous avons utilisé des anticorps hautement spécifiques des CPD et des 6-4PP, en conjonction avec la cytométrie en flux, afin de mettre au point une nouvelle technique permettant de quantifier précisément la cinétique de réparation de ces photoproduits en fonction du cycle cellulaire. Grâce à cette méthode, nous avons pu démontrer que dans les cellules humaines dont l’activité d’ATR est abrogée, la correction des dommages causés à l’ADN par la NER est complètement abolie durant la phase S. D’autre part, ATR ne joue aucun rôle dans la réparation de l’ADN pendant les phases G0/G1 ou G2/M [8]. Cette observation révèle une nouvelle fonction cruciale pour ATR dans le maintien de la stabilité génomique au cours de la réplication de l’ADN.
Nous avons en outre montré que sur six lignées tumorales sélectionnées aléatoirement, trois se caractérisent par une absence complète de la NER exclusivement au cours de la phase S. Il n’est donc pas exclu que de nombreuses tumeurs humaines puissent être caractérisées par un tel défaut. Ainsi, nos résultats sont d’une importance manifeste pour notre compréhension du développement des cancers, de même que pour leur traitement. En effet, le statut de la NER constitue un facteur de résistance clinique à certains agents chimiothérapeutiques couramment utilisés tels que le cisplatine [9] qui, à l’instar des UV, induit des lésions dans l’ADN réparées par la NER. Ainsi, les tumeurs présentant un défaut de la NER pendant la phase S pourraient répondre plus sélectivement à ces agents thérapeutiques, permettant ainsi l’élaboration de stratégies qui pourraient améliorer significativement l’efficacité des traitements du cancer.
Références
- Friedberg EC, Walker GC, Siede W, et al. DNA repair and mutagenesis. Washington DC : ASM Press, 2006. [Google Scholar]
- Melnikova VO, Ananthaswamy HN. Cellular and molecular events leading to the development of skin cancer. Mutat Res 2005; 571 : 91–106. [Google Scholar]
- Cleaver JE. Cancer in xeroderma pigmentosum and related disorders of DNA repair. Nat Rev Cancer 2005; 5 : 564–73. [Google Scholar]
- Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol 2008; 9 : 616–27. [Google Scholar]
- Tibbetts RS, Brumbaugh KM, Williams JM, et al. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev 1999; 13 : 152–7. [Google Scholar]
- Mathonnet G, Leger C, Desnoyers J, et al. UV wavelength-dependent regulation of transcriptioncoupled nucleotide excision repair in p53-deficient human cells. Proc Natl Acad Sci USA 2003; 100 : 7219–24. [Google Scholar]
- Matsuoka S, Ballif BA, Smogorzewska A, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007; 316 : 1160–6. [Google Scholar]
- Auclair Y, Rouget R, Affar el B, Drobetsky EA. ATR kinase is required for global genomic nucleotide excision repair exclusively during S phase in human cells. Proc Natl Acad Sci USA 2008; 105 : 17896–901. [Google Scholar]
- Olaussen KA, Dunant A, Fouret P, et al. DNA repair by ERCC1 in non-small-cell lung cancer and cisplatinbased adjuvant chemotherapy. N Engl J Med 2006; 355 : 983–91. [Google Scholar]
© 2009 médecine/sciences - Inserm / SRMS
Liste des figures
|
Figure 1. Réparation par excision de nucléotides. La NER procède selon deux sous-voies chevauchantes qui se distinguent uniquement à l’étape de reconnaissance de la lésion : (1) la réparation globale (global genomic NER, GG-NER) qui éradique des lésions dans l’ensemble du génome et (2) la réparation couplée à la transcription (transcription coupled NER ; TC-NER) qui agit uniquement sur le brin transcrit des gènes actifs. Lors de la GG-NER, via leurs capacités à déceler d’importantes déformations structurales, l’hétérodimère DDB1-DDB2 et/ou le complexe hétérotrimérique XPC-HR23B-CEN2 détectent les lésions. Alternativement, lors de la TC-NER, le blocage de l’ARN polymérase au site endommagé émet le signal à l’origine du recrutement des protéines CSA (Cockayne syndrome A protein) et CSB. À la suite de l’une ou l’autre de ces étapes de reconnaissance, la voie commune de la NER est mobilisée séquentiellement de la manière suivante : (1) les ADN hélicases (XPB et XPD), composantes du facteur de transcription TFIIH, déroulent la double hélice d’ADN ; (2) les protéines RPA (replication protein A) et XPA sont sollicitées afin de permettre la stabilisation du complexe et le recrutement des facteurs subséquents ; (3) l’arrivée des endonucléases XPG et XPF-ERCC1 mène à l’incision de la lésion en 3’ et 5’ respectivement et l’oligonucléotide endommagé (~20 bp) est excisé ; (4) l’ADN est resynthétisé par des facteurs de réplication, incluant les polymérases ε/δ, et finalement lié à nouveau par des ligases. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.