")
")
| Issue |
Med Sci (Paris)
Volume 24, Number 12, Décembre 2008
|
|
|---|---|---|
| Page(s) | 1077 - 1082 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/200824121077 | |
| Published online | 15 December 2008 | |
Sclérose latérale amyotrophique, jonction neuromusculaire et déficit énergétique
Amyotrophic lateral sclerosis: role of energy deficiency in neuromuscular junction dismantlement
Inserm U692, Laboratoire SMN, 67085 Strasbourg, France
Université Louis Pasteur, UMRS 692, Faculté de Médecine, 11, rue Humann, 67085 Strasbourg, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
La sclérose latérale amyotrophique (SLA) est la maladie du motoneurone de l’adulte la plus fréquente. Une partie des cas de SLA est liée à des mutations du gène codant la superoxyde dismutase à cuivre/zinc (SOD1) et l’analyse phénotypique de souris transgéniques exprimant des formes mutées de SOD1 (SOD1m) a permis de mieux comprendre les mécanismes physiopathologiques entraînant la mort du motoneurone. En revanche, l’extrapolation des résultats obtenus avec ces souris transgéniques s’est révélée décevante lors de plusieurs essais cliniques. Dans cette synthèse, nous résumons les principales données de la littérature concernant les mécanismes physiopathologiques à l’œuvre chez les souris SOD1m. En particulier, nous montrons comment en quelques années, les recherches ont mis en évidence que la degénérescence du motoneurone a lieu de façon rétrograde, via une déstabilisation initiale de la jonction neuromusculaire. La destruction des jonctions neuromusculaires est vraisemblablement la conséquence d’un déficit énergétique chronique au niveau de l’organisme entier. Ces résultats, associés à une comparaison entre le phénotype des souris SOD1m et celui des patients SLA, permettent d’envisager de nouvelles stratégies thérapeutiques et démontrent l’intérêt, mais aussi les limites, du modèle murin.
Abstract
Amyotrophic lateral sclerosis (ALS) is the most frequent adult onset motor neuron disorder. A subset of ALS cases is linked to mutations in the copper/zinc superoxide dismutase (sod1) gene and detailed phenotypic analysis of transgenic mice overexpressing mutant forms of SOD1 (mSOD1) allowed a better understanding of the pathophysiological mechanisms leading to motor neuron death. The promising results obtained in these animal models however poorly translated into conclusive clinical trials. In this review, we summarize the main pathological mechanisms at work in mSOD1 mice. In particular, recent results showed that the key pathological event was the destruction of the neuromuscular junction rather than motor neuron death. Neuromuscular junction dismantlement is likely the result of a chronic energy deficiency at the level of the whole organism. These results, along with a comparative analysis between the phenotype of mSOD1 mice and ALS patients, suggest new therapeutic strategies and show the interests but also the limits of the animal models.
© 2008 médecine/sciences - Inserm / SRMS
La sclérose latérale amyotrophique (SLA) est la maladie du motoneurone de l’adulte la plus répandue avec entre 1 et 3 nouveaux cas par an pour 100 000 personnes dans le monde. À ce titre, elle est la maladie neurodégénérative la plus fréquente après les maladies d’Alzheimer et de Parkinson. La SLA se manifeste par une paralysie progressive qui touche les muscles des membres (formes spinales) ou de la face (formes bulbaires). Le motoneurone inférieur, qui innerve les muscles squelettiques, dégénère et le motoneurone supérieur corticospinal est lui aussi affecté. La plupart des patients décèdent en 2 à 5 ans après le diagnostic, mais la maladie est très hétérogène tant dans sa durée que dans sa présentation clinique. Les options thérapeutiques sont quasi inexistantes. Ainsi, le riluzole1 reste à ce jour la seule molécule dont l’efficacité est prouvée, mais il n’augmente la survie des patients que de quelques mois.
La SLA se présente sous une forme familiale dans 20 % des cas, le plus souvent de transmission autosomique dominante, ou sous une forme sporadique car non associée à une histoire familiale connue. Il convient de noter que les formes familiales et sporadiques ne se distinguent pas sur le plan clinique. Au sein d’une même famille affectée, la présentation clinique de la maladie est hétérogène, ce qui souligne l’importance du contexte environnemental et suggère l’existence de gènes modulateurs de la pathologie. Plusieurs gènes, dont ceux codant pour l’angiogénine et la senataxine, et les gènes vapb (VAMP/synaptobrevin-associated membrane protein B) ou, plus récemment, tdp43 (TAR DNA-binding protein 43), ont été associés à des formes familiales, mais les mécanismes qui expliquent la dégénérescence sélective des motoneurones dans ces cas particuliers de SLA ne sont pas encore résolus [1, 2]. À l’inverse, le premier gène dont des mutations ont été liées à la SLA, le gène sod1, a, en revanche, été largement étudié. L’objectif de cette revue est de résumer les données les plus récentes concernant les mécanismes des formes à mutations SOD1 et d’exposer en quoi ces résultats s’appliquent aux autres formes de SLA.
Les souris porteuses de mutations de SOD1, unique modèle actuel de SLA
En 1993, Rosen et al. ont démontré que le locus als1, impliqué dans 20 % des cas familiaux de SLA (soit 1 à 2 % des cas totaux de SLA), était le gène codant la superoxyde dismutase à cuivre/zinc (SOD1) [3]. La SOD1 est une enzyme essentiellement cytosolique dont la fonction enzymatique est de dismuter l’anion superoxyde, l’un des sous-produits toxiques de la respiration mitochondriale, en peroxyde d’hydrogène (H2O2), lui-même détoxifié par la catalase. On connaît actuellement plus d’une centaine de mutations SOD1 et, de façon surprenante, il n’y a pas d’acide aminé ou de région de la protéine préférentiellement mutés.
La découverte des mutations SOD1 a permis la génération des premiers modèles animaux de SLA, les souris SOD1m. Ces souris surexpriment une forme mutée de SOD1 et développent une maladie du motoneurone. Il convient de noter que la surexpression d’une SOD1 mutée, mais pas celle de la forme sauvage, provoque une SLA chez la souris. De plus, l’ablation du gène sod1 n’a que très peu d’effets. Cela suggère que les protéines SOD1m acquièrent une nouvelle fonction toxique pour le motoneurone (la notion de gain de fonction de la SOD1m a fait récemment l’objet d’excellentes revues et nous ne la discuterons pas plus en détails [1, 2]). D’autres lignées de souris ont été créées pour modéliser la SLA à l’aide de gènes candidats mais les résultats se sont avérés décevants. La découverte récente de mutations du gène tdp43 devrait enfin aboutir à la création de nouveaux modèles murins. Ainsi, et malgré toutes les réserves qui ont été émises sur ces souris SOD1m (voir ci-dessous), elles développent un phénotype ressemblant à la SLA et restent notre seul modèle animal actuel et notre unique ressource pour les études précliniques.
La déstabilisation de la jonction neuromusculaire est l’événement pathologique primaire
Au cours des années 1990 et 2000, la plupart des travaux ont eu pour objectif de mieux comprendre l’évolution de la pathologie chez les souris SOD1m. À l’âge adulte, les souris SOD1m développent une faiblesse musculaire associée à une dénervation musculaire massive et à une dégénérescence des motoneurones inférieurs et supérieurs. Le premier événement semble être la destruction de la jonction neuromusculaire, en particulier de la structure post-synaptique. La dégradation de l’axone du motoneurone puis la destruction du corps cellulaire surviennent ensuite [4–6]. Pendant très longtemps, il paraissait prometteur de trouver une stratégie thérapeutique qui permette de sauver le corps cellulaire du motoneurone de l’apoptose, ce qui préserverait les chances de régénération de la jonction neuromusculaire.
Ainsi, les très nombreux travaux étudiant les mécanismes de la mort du corps cellulaire du motoneurone ont clairement établi que celle-ci, chez l’homme comme chez la souris SOD1m, est de type apoptotique, et que de multiples voies de transduction participent in vivo à la dégénérescence du corps cellulaire (pour revue, voir [7]). Sur le plan thérapeutique, ces travaux se sont révélés décevants. En effet, l’inhibition pharmacologique ou génétique des voies d’apoptose n’a que peu d’effets sur la survie des souris SOD1m [7]. De plus, trois études récentes ont montré que la protection du corps cellulaire du motoneurone, et donc la préservation du nombre de corps cellulaires de motoneurones dans la moelle épinière, ne permettaient pas de guérir la souris SOD1m [8–10] : l’ablation de bax, un gène clé de l’apoptose dans le motoneurone, abolit complètement l’apoptose du motoneurone mais ne retarde que modestement la mort de l’animal et la dénervation musculaire [9]. De même, le valproate de sodium, qui inhibe le remodelage de la chromatine lors de l’apoptose du motoneurone, ou un inhibiteur de P38 MAPK, une protéine kinase intervenant dans les étapes initiales de l’apoptose, sont capables de protéger le corps cellulaire du motoneurone, mais ne retardent pas ou peu la dénervation musculaire et la mort de l’animal [8, 10].
Comment interpréter ces résultats ? L’événement pathologique primaire, qui détermine la mort de l’animal, ne semble pas être la mort du corps cellulaire du motoneurone, mais la disparition des jonctions neuromusculaires. De ce fait, les motoneurones protégés pharmacologiquement ou génétiquement sont inutiles car ils n’arrivent pas à recréer les jonctions neuromusculaires précédemment détruites. La mort par apoptose du corps cellulaire serait donc une conséquence secondaire de cette destruction synaptique (Figure 1).
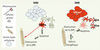 |
Figure 1. Mécanismes de la SLA : évolution des modèles entre 2000 et 2008. En 2000, l’hypothèse prédominante était que la SOD1m exprimée dans le motoneurone provoquait l’apoptose de cette cellule et, par conséquent, la dénervation du muscle, la paralysie progressive et la mort. Les travaux les plus récents ont montré que le site de toxicité principal de la SOD1m n’était pas le neurone, mais plusieurs types cellulaires dont les différentes cellules gliales. L’expression musculaire de la SOD1m ne semble pas impliquée. De plus, il semble que la destruction de la jonction neuromusculaire (JNM) soit l’événement déterminant de la survie de l’animal. Il reste à démontrer que le déficit énergétique musculaire est par lui-même capable de déstabiliser la jonction neuromusculaire et à comprendre les mécanismes provoquant ce déficit énergétique musculaire. En rouge : cellule où l’expression de la SOD1m est critique pour la pathogénie. |
Quel est le site de la toxicité de la SOD1m ?
Ces travaux suggéraient que la place centrale attribuée à la mort du motoneurone, étape finale indiscutable de la pathologie, était largement surestimée. Une deuxième série d’études est venue compléter cette notion et bousculer définitivement les paradigmes existants.
La SOD1 est une enzyme ubiquitaire et la plupart des travaux exploraient l’hypothèse selon laquelle la SOD1m déclenchait la cascade apoptotique dans le motoneurone lui même, par exemple en interagissant directement avec la machinerie apoptotique. Au contraire, il est maintenant clairement établi que l’expression de la SOD1m dans le motoneurone n’a qu’une importance marginale dans le déclenchement de la maladie. Ainsi, l’expression ciblée de la SOD1m dans le motoneurone ne suffit pas à provoquer la maladie chez la souris [11, 12]. En accord avec ces résultats, la démarche expérimentale symétrique, qui consiste à diminuer l’expression de la SOD1 mutée dans le motoneurone via un système de recombinaison homologue, n’augmente que peu la survie des souris SOD1m [13]. Finalement, la création de chimères entre souris SOD1m et sauvages a montré qu’un motoneurone porteur de la mutation SOD1 pouvait survivre dans un environnement riche en cellules sauvages tandis qu’un motoneurone sauvage, placé dans un environnement riche en cellules SOD1m, pouvait développer des marqueurs histopathologiques de souffrance comme les agrégats d’ubiquitine [14, 15]. De fait, le destin d’un motoneurone ne semble que peu lié à son expression endogène de SOD1m. La SOD1m exerce donc sa toxicité dans d’autres cellules et cela provoque secondairement la destruction de la jonction neuromusculaire puis la mort du motoneurone.
Plusieurs types cellulaires pourraient participer à la toxicité des SOD1m. Parmi les candidats, se trouvent les cellules gliales (astrocytes, microglie, cellules de Schwann) ou les cellules musculaires [16]. Ainsi, le laboratoire de Luis Barbeito a montré que les astrocytes porteurs d’une SOD1m sont capables de provoquer l’apoptose de motoneurones sauvages via un mécanisme impliquant le stress oxydant et le NGF (nerve growth factor) [17, 18]. Ces travaux ont récemment été confirmés par d’autres [19, 20] et suggèrent que l’astrocyte pourrait participer à la toxicité envers le motoneurone. À l’appui de cette hypothèse, la diminution de l’expression de la SOD1 mutée spécifiquement dans les astrocytes retarde la progression des symptômes [21]. Un deuxième type cellulaire, les macrophages, dont la microglie est un sous-type, est susceptible de contribuer à la pathologie. Ainsi, comme pour les astrocytes, diminuer, par recombinaison ciblée, l’expression de la SOD1m dans les cellules de la lignée macrophage/microglie [13], ou greffer des macrophages sauvages chez les souris SOD1m, augmente significativement la durée de vie des animaux [22]. Il n’en reste pas moins que ni l’expression astrocytaire ni l’expression microgliale seules de la SOD1m ne sont capables de provoquer un phénotype de SLA. D’autres cellules, notamment les cellules musculaires ou les cellules de Schwann, sont aussi potentiellement impliquées dans la toxicité de la SOD1m, mais leur rôle est soit controversé, soit encore inexploré.
Le muscle est-il l’initiateur de la destruction de la jonction neuromusculaire ?
Le rôle pathologique primaire de la disparition de la jonction neuromusculaire suggère que le muscle pourrait jouer un rôle important dans l’initiation de la maladie. En effet, des données de notre laboratoire sont en accord avec cette hypothèse. Nous avions montré que la protéine Nogo-A, connue pour son rôle d’inhibiteur de la croissance neuritique dans le système nerveux central, était massivement surexprimée dans le muscle squelettique de souris SOD1m et de patients atteints de SLA [23]. Cette surexpression est limitée aux fibres oxydatives (type I) chez le patient et les niveaux musculaires de Nogo-A sont corrélés avec l’état clinique du patient [24]. Cela nous a d’ailleurs permis de proposer la détection de cette protéine comme marqueur diagnostique dans certaines formes de SLA [25]. De plus, l’ablation de Nogo-A augmente la durée de vie des souris SOD1m, tandis que sa surexpression musculaire détruit les jonctions neuromusculaires [26]. À partir de l’exemple de Nogo-A, il semble clair que des modifications de l’expression musculaire de certains gènes sont capables de moduler la pathologie développée par les souris SOD1m. Cela est cohérent avec l’efficacité de certains traitements spécifiquement ciblés vers le muscle, comme l’IGF1 (insulin growth factor 1) [27]. Cependant, deux études suggèrent que l’expression de la SOD1m au niveau musculaire n’est pas déterminante dans la pathologie [28, 29]. Il est ainsi parfaitement possible que la toxicité musculaire, via Nogo-A par exemple, soit provoquée indirectement par l’expression de la SOD1m dans un autre type cellulaire. Ainsi, de très nombreuses cytokines sécrétées par les macrophages sont capables d’agir sur l’expression des gènes musculaires et l’expression macrophagique de la SOD1m pourrait être la cause de la toxicité musculaire envers le motoneurone.
Les modèles murins de SLA présentent des altérations marquées du métabolisme énergétique
Comment la destruction de la jonction neuromusculaire est-elle déclenchée ? D’autres travaux de notre laboratoire suggèrent que des modifications du métabolisme musculaire pourraient en être la cause directe. Notre observation initiale était que les souris SOD1m avaient un déficit pondéral par rapport aux souris sauvages. Ce déficit énergétique n’est pas causé par une diminution de la prise alimentaire, mais par une augmentation du métabolisme basal et le métabolisme énergétique, en particulier lipidique, est extrêmement perturbé chez les souris SOD1m [30, 31]. De plus, le profil d’expression des gènes dans le muscle ainsi que l’augmentation de la capture de glucose par le muscle suggèrent une augmentation du métabolisme énergétique dans ce tissu. Ainsi, chez la souris SOD1m, le métabolisme musculaire augmenté provoque une fonte des réserves adipeuses et un hypermétabolisme de l’animal, et entraîne un déficit énergétique chronique. Ces anomalies très précoces précèdent l’amyotrophie et les défauts électromyographiques, et n’en sont donc pas une conséquence.
Le déficit énergétique chronique et l’hypermétabolisme musculaire contribuent-ils à la maladie du motoneurone ? Pour le savoir, nous avons augmenté la ration calorique des souris SOD1m en les nourrissant avec un régime enrichi en lipides. Cette manipulation nutritionnelle a corrigé le déficit énergétique et nous avons observé une nette augmentation de la survie des animaux. De plus, la survie des motoneurones était augmentée et la dénervation musculaire diminuée. Cet effet protecteur du régime hyperlipidique est dose-dépendant puisque des données récentes du laboratoire de Mark Mattson confirment nos résultats et montrent qu’une ration énergétique plus élevée que celle utilisée dans nos conditions expérimentales est encore plus efficace en terme d’accroissement de la survie des souris SOD1m [32]. Les souris SOD1m sont donc en déficit énergétique et la correction nutritionnelle de ce déficit améliore le phénotype clinique. À l’inverse, aggraver le déficit énergétique, en soumettant les souris SOD1m à une restriction calorique, exacerbe le phénotype clinique, ce qui confirme que le déficit énergétique est un déterminant important de la pathologie de ces souris. Ces études semblent distinguer la SLA des autres maladies neurodégénératives. En effet, les modèles transgéniques de maladie d’Alzheimer ou de Huntington ont un phénotype métabolique opposé à celui des souris SOD1m, avec une tendance à l’obésité et à l’insulino-résistance, et sont protégés par la restriction calorique [33, 34] et aggravés par le régime hyperlipidique. Les causes de ces différences entre maladies neurodégénératives sont à l’heure actuelle inconnues, mais leur élucidation pourrait amener à comprendre les bases de la sélectivité neuronale de ces pathologies.
Métabolisme énergétique chez les patients SLA : points communs et différences avec les souris SOD1m
Le déficit énergétique et l’hypermétabolisme existent-ils chez les patients ? Plusieurs études du Pr Couratier avaient montré que les patients SLA étaient, tout comme les souris SOD1m, hypermétaboliques [35–37], ce qui suggérait une certaine conservation des mécanismes pathologiques. Afin de déterminer si ces altérations du métabolisme énergétique étaient aussi retrouvées chez les patients, nous avons comparé les taux de lipides circulants chez plusieurs centaines de patients SLA et de patients contrôles. Nous avons observé que les patients ayant une SLA de type sporadique présentaient des niveaux anormalement élevés de lipides circulants [38]. Ainsi, dans notre étude, près d’un patient SLA sur 2 a des niveaux de LDL-cholestérol considérés comme trop élevés. De plus, les patients SLA présentant un rapport LDL/HDL2 anormalement élevé, facteur de risque cardiovasculaire bien caractérisé, avaient une survie plus élevée de 15 mois, signifiant donc que la dyslipidémie est un facteur protecteur dans la SLA. En conclusion, souris SOD1m et patients SLA développent une maladie métabolique, avec un hypermétabolisme et un déficit énergétique. Les détails du tableau clinique sont cependant différents, ainsi, les patients SLA sont hyperlipidémiques, alors que les souris SOD1m ont une tendance marquée à l’hypolipidémie (Tableau I). Cependant, dans les deux cas, l’augmentation des lipides circulants ralentit la progression de la maladie. À l’heure actuelle, les arguments expérimentaux chez les patients SLA sont purement corrélatifs et la correction du déficit énergétique chez les patients SLA une piste thérapeutique encore inexplorée. Nos résultats suggèrent déjà que l’utilisation des agents hypolipémiants comme les statines chez les patients SLA pourrait être néfaste.
Phénotype métabolique comparé des patients SLA et des souris SOD1m.
Lost in translation : les échecs de l’utilisation préclinique des souris SOD1m
De nombreux composés ont montré une efficacité dans le modèle murin. Il paraissait donc raisonnable d’envisager un essai clinique avec les molécules les plus efficaces. La litanie des essais cliniques infructueux a de ce fait été particulièrement décourageante : la créatine, l’amoxyfilline, la minocycline… ont toutes montré des effets bénéfiques chez la souris SOD1m. Aucun de ces composés n’a cependant d’efficacité chez l’homme. Pire, certains aggravent au contraire la maladie… (Tableau II) [39].
Quelles peuvent être les raisons de ces échecs ? Une première explication pourrait être liée à l’extrapolation des résultats obtenus entre un modèle murin de forme familiale à la population globale de patients SLA. En effet, s’il est très probable que les mécanismes identifiés chez les souris SOD1m soient valables chez les patients porteurs d’une mutation SOD1 (1 à 2 % des cas), rien ne prouve que ce soit le cas de l’ensemble des cas de SLA. Il ne peut donc être exclu qu’il faille traiter différemment les patients porteurs d’une SOD1 mutée et les autres, mais la réponse à une telle question sera certainement quasi impossible à obtenir étant donné la lourdeur des essais cliniques et la faible proportion de patients porteurs de mutations SOD1. Une deuxième explication est aussi envisageable : les traitements chez la souris sont généralement donnés avant la phase symptomatique, ce qui est impossible chez le patient. Il est possible que les essais cliniques soient réalisés avec des patients dont la pathologie est trop avancée pour que la molécule soit efficace. Enfin, peut-être avons-nous oublié trop vite que les souris SOD1m sont avant tout… des souris, c’est-à-dire un mammifère avec certaines caractéristiques, en particulier métaboliques, très différentes des primates [41]. Des différences fines d’espèce pourraient expliquer des résultats complètement différents chez les patients, et c’est particulièrement vrai du point de vue du métabolisme énergétique. Un exemple simple illustre cet aspect : les muscles de souris sont essentiellement glycolytiques, et donc utilisateurs de créatine phosphate, ce qui n’est pas le cas des muscles humains, plus oxydatifs. Une telle différence pourrait expliquer l’efficacité de la créatine dans le modèle murin, et son inefficacité chez le patient. Il semble donc qu’entre des résultats précliniques encourageants chez la souris SOD1m et l’essai clinique, il faille réaliser des études cliniques intermédiaires et créer de nouveaux modèles de SLA chez des espèces plus proches de l’homme comme les primates non humains pour valider un mécanisme observé chez la souris.
Approches thérapeutiques de la souris SOD1m au patient SLA : une litanie d’échecs.
Conclusion
Près de quinze ans après la création des premières souris SOD1m, nous avons beaucoup progressé dans la compréhension des mécanismes de la SLA. D’une maladie restreinte au motoneurone, la SLA est devenue une pathologie systémique dont la conséquence ultime est la disparition des motoneurones. Une analyse comparée des pathologies chez la souris et les patients pourra apporter dans les prochaines années des pistes thérapeutiques nouvelles, qu’elles soient pharmacologiques, nutritionnelles ou autre.

Remerciements
L’unité Inserm U692 est soutenue par l’Association française de lutte contre les myopathies (AFM), l’Association de recherche sur la SLA (ARS), la Fédération de recherches sur le cerveau (FRC), la Fondation recherche médiacle (FRM) et l’Association pour la recherche et le développement de moyens de lutte contre les maladies neurodégénératives (AREMANE).
Riluzole : agit en bloquant les canaux sodiques dépendant du voltage et en diminuant ainsi la libération présynaptique du glutamate.
LDL : low density lipoproteins ; HDL : high density lipoproteins.
Références
- Gonzalez de Aguilar JL, Echaniz-Laguna A, Fergani A, et al. Amyotrophic lateral sclerosis: all roads lead to Rome. J Neurochem 2007; 101 : 1153–60. [Google Scholar]
- Boillee S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 2006; 52 : 39–59. [Google Scholar]
- Rosen DR, Siddique T, Patterson D, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993; 362 : 59–62. [Google Scholar]
- Fischer LR, Culver DG, Tennant P, et al. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol 2004; 185 : 232–40. [Google Scholar]
- Frey D, Schneider C, Xu L, et al. Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J Neurosci 2000; 20 : 2534–42. [Google Scholar]
- Pun S, Santos AF, Saxena S, et al. Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat Neurosci. 2006; 9 : 408–19. [Google Scholar]
- Guegan C, Przedborski S. Programmed cell death in amyotrophic lateral sclerosis. J Clin Invest 2003; 111 : 153–61. [Google Scholar]
- Rouaux C, Panteleeva I, Rene F, et al. Sodium valproate exerts neuroprotective effects in vivo through CREB-binding protein-dependent mechanisms but does not improve survival in an amyotrophic lateral sclerosis mouse model. J Neurosci 2007; 27 : 5535–45. [Google Scholar]
- Gould TW, Buss RR, Vinsant S, et al. Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS. J Neurosci 2006; 26 : 8774–86. [Google Scholar]
- Dewil M, De la Cruz VF, Van Den Bosch L, et al. Inhibition of p38 mitogen activated protein kinase activation and mutant SOD1 (G93A)-induced motor neuron death. Neurobiol Dis 2007; 26 : 332–41. [Google Scholar]
- Pramatarova A, Laganiere J, Roussel J, et al. Neuron-specific expression of mutant superoxide dismutase 1 in transgenic mice does not lead to motor impairment. J Neurosci 2001; 21 : 3369–74. [Google Scholar]
- Lino MM, Schneider C, Caroni P. Accumulation of SOD1 mutants in postnatal motoneurons does not cause motoneuron pathology or motoneuron disease. J Neurosci 2002; 22 : 4825–32. [Google Scholar]
- Boillee S, Yamanaka K, Lobsiger CS, et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science 2006; 312 : 1389–92. [Google Scholar]
- Clement AM, Nguyen MD, Roberts EA, et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 2003; 302 : 113–7. [Google Scholar]
- Pettmann B, Raoul C, Haase G. Mort des motoneurones dans la SLA : suicide ou meurtre ? Med Sci (Paris) 2006; 22 : 923–5. [Google Scholar]
- Boillée S, Lobsiger CS. Les cellules gliales : pas d’un si grand support pour les motoneurones. Med Sci (Paris) 2008; 24 : 124–6. [Google Scholar]
- Pehar M, Cassina P, Vargas MR, et al. Astrocytic production of nerve growth factor in motor neuron apoptosis: implications for amyotrophic lateral sclerosis. J Neurochem 2004; 89 : 464–73. [Google Scholar]
- Vargas MR, Pehar M, Cassina P, et al. Increased glutathione biosynthesis by Nrf2 activation in astrocytes prevents p75NTR-dependent motor neuron apoptosis. J Neurochem 2006; 97 : 687–96. [Google Scholar]
- Nagai M, Re DB, Nagata T, et al. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci 2007; 10 : 615–22. [Google Scholar]
- Di Giorgio FP, Carrasco MA, Siao MC, et al. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat Neurosci 2007; 10 : 608–14. [Google Scholar]
- Yamanaka K, Chun SJ, Boillee S, et al. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci 2008; 11 : 251–3. [Google Scholar]
- Beers DR, Henkel JS, Xiao Q, et al. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci USA 2006; 103 : 16021–6. [Google Scholar]
- Dupuis L, Gonzalez de Aguilar JL, di Scala F, et al. Nogo provides a molecular marker for diagnosis of amyotrophic lateral sclerosis. Neurobiol Dis 2002; 10 : 358–65. [Google Scholar]
- Jokic N, Gonzalez de Aguilar JL, Pradat PF, et al. Nogo expression in muscle correlates with amyotrophic lateral sclerosis severity. Ann Neurol 2005; 57 : 553–6. [Google Scholar]
- Pradat PF, Bruneteau G, Gonzalez de Aguilar JL, et al. Muscle Nogo-A expression is a prognostic marker in lower motor neuron syndromes. Ann Neurol 2007; 62 : 15–20. [Google Scholar]
- Jokic N, Gonzalez de Aguilar JL, Dimou L, et al. The neurite outgrowth inhibitor Nogo-A promotes denervation in an amyotrophic lateral sclerosis model. EMBO Rep 2006; 7 : 1162–7. [Google Scholar]
- Dobrowolny G, Giacinti C, Pelosi L, et al. Muscle expression of a local Igf-1 isoform protects motor neurons in an ALS mouse model. J Cell Biol 2005; 168 : 193–9. [Google Scholar]
- Miller TM, Kim SH, Yamanaka K, et al. Gene transfer demonstrates that muscle is not a primary target for non-cell-autonomous toxicity in familial amyotrophic lateral sclerosis. Proc Natl Acad Sci USA 2006; 103 : 19546–51. [Google Scholar]
- Towne C, Raoul C, Schneider BL, et al. Systemic AAV6 delivery mediating RNA interference against SOD1: neuromuscular transduction does not alter disease progression in fALS mice. Mol Ther 2008; 16 : 1018–25. [Google Scholar]
- Dupuis L, Oudart H, Rene F, et al. Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: benefit of a high-energy diet in a transgenic mouse model. Proc Natl Acad Sci USA 2004; 101 : 11159–64. [Google Scholar]
- Fergani A, Oudart H, Gonzalez De Aguilar JL, et al. Increased peripheral lipid clearance in an animal model of amyotrophic lateral sclerosis. J Lipid Res 2007; 48 : 1571–80. [Google Scholar]
- Mattson MP, Cutler RG, Camandola S. Energy intake and amyotrophic lateral sclerosis. Neuromolecular Med 2007; 9 : 17–20. [Google Scholar]
- Maswood N, Young J, Tilmont E, et al. Caloric restriction increases neurotrophic factor levels and attenuates neurochemical and behavioral deficits in a primate model of Parkinson’s disease. Proc Natl Acad Sci USA 2004; 101 : 18171–6. [Google Scholar]
- Rasouri S, Lagouge M, Auwerx J. SIRT1/PGC-1: un axe neuroprotecteur ? Med Sci (Paris) 2007; 23 : 840–4. [Google Scholar]
- Desport JC, Preux PM, Magy L, et al. Factors correlated with hypermetabolism in patients with amyotrophic lateral sclerosis. Am J Clin Nutr 2001; 74 : 328–34. [Google Scholar]
- Desport JC, Preux PM, Truong TC, et al. Nutritional status is a prognostic factor for survival in ALS patients. Neurology 1999; 53 : 1059–63. [Google Scholar]
- Desport JC, Torny F, Lacoste M, et al. Hypermetabolism in ALS: correlations with clinical and paraclinical parameters. Neurodegenerative Dis 2005; 2 : 202–7. [Google Scholar]
- Dupuis L, Corcia P, Fergani A, et al. Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology 2008; 70 : 1004–9. [Google Scholar]
- Benatar M. Lost in translation: treatment trials in the SOD1 mouse and in human ALS. Neurobiol Dis 2007; 26 : 1–13. [Google Scholar]
- Dupuis L, Di Scala F, Rene F, et al. Up-regulation of mitochondrial uncoupling protein 3 reveals an early muscular metabolic defect in amyotrophic lateral sclerosis. FASEB J 2003; 17 : 2091–93. [Google Scholar]
- Langui D, Lachapelle F, Duyckaerts C. Modèles animaux des maladies neuro-dégénératives. Med Sci (Paris) 2007; 23 : 180–6. [Google Scholar]
- Dobrowolny G, Aucello M, Rizzuto E, et al. Skeletal muscle is a primary target of SOD1G93A mediated toxicity. Cell Metab 2008; 8 : 425–36. [Google Scholar]
Liste des tableaux
Approches thérapeutiques de la souris SOD1m au patient SLA : une litanie d’échecs.
Liste des figures
|
Figure 1. Mécanismes de la SLA : évolution des modèles entre 2000 et 2008. En 2000, l’hypothèse prédominante était que la SOD1m exprimée dans le motoneurone provoquait l’apoptose de cette cellule et, par conséquent, la dénervation du muscle, la paralysie progressive et la mort. Les travaux les plus récents ont montré que le site de toxicité principal de la SOD1m n’était pas le neurone, mais plusieurs types cellulaires dont les différentes cellules gliales. L’expression musculaire de la SOD1m ne semble pas impliquée. De plus, il semble que la destruction de la jonction neuromusculaire (JNM) soit l’événement déterminant de la survie de l’animal. Il reste à démontrer que le déficit énergétique musculaire est par lui-même capable de déstabiliser la jonction neuromusculaire et à comprendre les mécanismes provoquant ce déficit énergétique musculaire. En rouge : cellule où l’expression de la SOD1m est critique pour la pathogénie. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.