")
")
| Numéro |
Med Sci (Paris)
Volume 24, Numéro 4, Avril 2008
|
|
|---|---|---|
| Page(s) | 399 - 406 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/2008244399 | |
| Publié en ligne | 15 avril 2008 | |
Récepteur de la ryanodine et dysfonctionnement myocardique
Ryanodine receptor and heart disease
Inserm, U 637, F-34925 Montpellier, France.
Université Montpellier1, UFR de Médecine, CHU Arnaud de Villeneuve, 34295 Montpellier Cedex 5, France
Le calcium (Ca2+) joue un rôle essentiel dans la contraction cardiaque. Il est stocké dans le réticulum sarcoplasmique (RS) et libéré massivement à chaque battement cardiaque par le canal calcique du RS : le récepteur de la ryanodine de type 2 (RyR2). Ce canal est un homotétramère dont chacune des sous-unités, formant le pore, est associée à des protéines régulatrices telles que la calstabine 2, la calmoduline, la PKA, la CamKII, ou encore la calsequestrine, pour former un complexe macromoléculaire qui joue un rôle déterminant au cours de pathologies cardiaques. La modification de l’activité du canal ou de l’une de ses protéines régulatrices conduit à des troubles du rythme parfois fatals. La découverte d’une pharmacologie spécifique du RyR2 présente par conséquent un enjeu thérapeutique majeur.
Abstract
Calcium ions (Ca2+) play an essential role in cardiac excitation-contraction coupling. Ca2+ is stored in the sarcoplasmic reticulum (SR) and is release via SR-Carelease channels (ryanodine receptors, RyR2) to trigger contraction. RyR2 is a homotetramer comprising 4 poreforming subunits. Each subunit is closely associated to regulatory proteins such as calstabine 2 (FKBP12.6), calmodulin, PKA, CamKII, calsequestrin and form a macromolecular complex that plays a critical role in pathological conditions. As a matter of fact, alterations of the channel activity and/or associated regulatory proteins can cause severe functional alterations resulting in arrhythmias and sudden death. Thus, RyR2 represent a novel therapeutic target and the discovery of a new pharmacological agent able to restore a normal RyR2 channel function represents a major challenge in the cardiac field.
© 2008 médecine/sciences - Inserm / SRMS
Le couplage excitation-contraction (CEC) est un mécanisme fondamental, caractéristique des cellules contractiles musculaires squelettiques et cardiaques. Il désigne l’ensemble des processus qui assurent la transformation du stimulus électrique (potentiel d’action, PA) en un signal intracellulaire permettant d’activer la contraction des myocytes. La dépolarisation membranaire conduit à l’ouverture des canaux calciques de type L (aussi appelés récepteurs des dihydropyridines, DHPR) situés essentiellement sur des invaginations membranaires appelées tubules transverses. L’entrée de Ca2+ qui en résulte est insuffisante pour déclencher la contraction. Elle est cependant suffisante pour activer l’ouverture du canal calcique du réticulum sarcoplasmique (RS), aussi appelé récepteur de la ryanodine, RyR, selon un mécanisme autocatalytique appelé CICR (Ca2+-induced Ca2+-release). Ainsi, la concentration en Ca2+ ([Ca2+]) cytoplasmique passe localement d’une valeur de repos de ~100 nM à 1 µM, suffisante pour activer les protéines contractiles et la contraction musculaire. Cette fonction cellulaire hautement spécialisée, qui permet de réguler les variations intracellulaires de la [Ca2+], repose sur une architecture membranaire, une compartimentation cellulaire et une organisation moléculaire parfaitement adaptées qui placent le RyR au centre du CEC en situation physiologique ou pathologique.
Récepteur de la ryanodine : un complexe macromoléculaire
Trois isoformes de RyR (RyR1, RyR2, RyR3) ont été identifiées et leurs gènes clonés. Elles sont codées par 3 gènes différents et présentent 70 % d’homologies entre elles [1]. Seule la protéine RyR2 est exprimée dans le cœur. C’est un récepteur homotétramérique (565 kDa par sous-unité). L’association quadrangulaire des sous-unités dessine un pore central permettant la diffusion des ions Ca2+. La structure tridimensionnelle complète de cette macromolécule n’est pas encore connue. On sait cependant que chaque sous-unité est formée d’une partie transmembranaire carboxy-terminale formant le pore du canal et d’une large partie cytoplasmique amino-terminale appelée « pied », impliquée dans la régulation de l’activité du RyR2 par une interaction de type protéine-protéine (Figure 1) [2]. Chacune des sous unités est associée à des protéines régulatrices pour former un complexe macromoléculaire. L’intégrité de leur interaction avec les sous unités du RyR2 joue un rôle déterminant au cours de pathologies cardiaques. Les principales protéines régulatrices caractérisées à ce jour sont décrites ci-dessous.
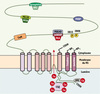 |
Figure 1. Représentation schématique du complexe macromoléculaire formé par le récepteur de la ryanodine et ses protéines associées. CaM : calmoduline ; CSQ : calsequestrine ; FKBP : calstabine 2. |
La calstabine 2 (FKBP12.6) est une enzyme à activité peptidyl-propyl-cis-trans-isomérase de 12,6 kDa qui se fixe sur chaque monomère de RyR2 avec une forte affinité et une stœchiométrie de 1:1. Les mesures électrophysiologiques de l’activité des RyR2 montrent que l’interaction de la calstabine 2 avec le RyR2 stabilise l’état fermé du canal ; sa dissociation augmente la probabilité d’ouverture (Po) et induit des états de sous-conductance [3]. La calstabine 2 est certainement la protéine régulatrice qui a suscité à ce jour le plus grand intérêt sur le plan mécanistique, en raison de son rôle important en physiopathologie.
La calmoduline est une protéine ubiquitaire de 17 kDa à haute affinité pour le Ca2+, qui peut se fixer avec une stœchiométrie de 1:1 sur chaque monomère de RyR2. Elle exerce une action inhibitrice sur RyR2 quelle que soit la [Ca2+] [4]. Par ailleurs, l’interaction RyR2/calmoduline dépend fortement de l’état Redox des milieux. Une déficience en calmoduline conduit, chez la souris, à une hypertrophie myocardique et à une mort prématurée [5]. Une telle dysfonction n’a cependant pas encore été observée chez l’homme.
Le RyR2 est également associé à la protéine kinase A (PKA) et la Ca2+/calmoduline Kinase II (CamKII). La stimulation β-adrénergique entraîne la phosphorylation du RyR2 par la PKA sur la sérine 2808, ce qui conduit à la dissociation transitoire de la calstabine 2 et à l’activation du canal. C’est un effet adaptatif bénéfique, en réponse à un stress, qui permet une meilleure performance du CEC. Cependant, une hyperphosphorylation chronique du RyR2 par la PKA a un effet délétère qui maintient le canal dans un état de sous-conductance permanent. La PKA est liée au RyR2 par la protéine d’ancrage mAKAP qui assure également une interaction étroite avec la phosphodiestérase de type 4D3 (PDE4D3) [6]. Les phosphatases de type 1 et 2A (PP2A) sont également situées à proximité. Elles sont liées au RyR2 par leurs protéines d’ancrage respectives, la spinophiline et la PR130. Les phosphatases et les phosphodiestérases permettent, respectivement, de déphosphoryler le canal et de réduire localement la concentration en AMPc.
Le RyR2 peut également être phosphorylé sur la sérine 2815 par la CamKII. Cette phosphorylation, qui n’est pas associée à une dissociation de calstabine 2, augmente la Po du RyR2 [7], de manière plus modérée que la phosphorylation PKA-dépendante [8]. Elle semble jouer un rôle important dans la régulation du CEC cardiaque lors de changements du rythme et serait impliquée dans le contrôle de la relation force-fréquence [8]. Certains auteurs lui accordent cependant un rôle possible au cours de l’insuffisance cardiaque
La sorcine est une protéine cytosolique de 22 kDa, à haute affinité pour le Ca2+, qui se lie également au RyR2. Elle exerce une action inhibitrice sur le RyR2. Cet effet est réduit lorsque la sorcine est phosphorylée par la PKA [9, 10].
Les protéines évoquées ci-dessus exercent leur action sur le RyR2 par une interaction sur la boucle cytoplasmique. On dénombre cependant d’importantes régulations qui s’opèrent via la face luminale ou face interne de la membrane du RS. Cette régulation fait, en particulier, intervenir des protéines à fort pourvoir chélateur du Ca2+ telles que la calséquestrine ou les HRC (histidine rich Ca2+-binding protein) qui permettent de maintenir une forte [Ca2+] dans le RS sous forme liée et sous forme libre (~1 mM). La calséquestrine interagit directement avec le RyR2 et module son activité. Par ailleurs, une réduction importante de la [Ca2+] du RS conduit à la fermeture des RyR2 et à une période réfractaire qui se maintient jusqu’à la reconstitution des stocks calciques du RS par la Ca2+ ATPase du RS (SERCA) [11]. On recense enfin d’autres protéines transmembranaires interagissant avec le RyR2 telles que la triadine, la junctine, la junctate et les junctophilines qui semblent aussi moduler son activité. Le rôle précis de chacune de ces molécules reste à explorer et cette liste est, par ailleurs, certainement loin d’être exhaustive.
Régulation des RyR2
Le RyR2, du fait de sa structure macromoléculaire complexe, présente différents niveaux de régulation de l’activité du canal (Tableau I), avec un rôle prépondérant des phosphorylations contrôlées de façon directe par l’activité des kinases (PKA et CamKII), et des phosphatases et phosphodiestérases. Cependant ces RyR2 sont également régulés par différentes « petites molécules » et facteurs régulateurs non protéiques essentiels.
Liste des principales protéines régulatrices associées au récepteur de la ryanodine et formant le complexe macromoléculaire.
Régulation cationique
Le RyR2 porte sur le pied deux sites de fixation pour le Ca2+ dont l’un, de haute affinité, est responsable de l’activation et de l’ouverture du canal ; l’autre site, de faible affinité, est responsable de l’inactivation du canal. Ces deux sites peuvent également fixer du Mg2+ qui exerce, en revanche, une activité inhibitrice sur les deux sites. Dans les conditions normales, la concentration en Mg2+ est relativement stable [12]. Ainsi, le Ca2+ est le seul cation divalent à exercer un rôle fonctionnel important. Le RyR2 est également décrit comme sensible aux protons et, par conséquent, au pH. Bien que le rôle du pH soit controversé, ses effets se manifesteraient pour des acidifications importantes comme au cours d’une ischémie. Il semble également que l’effet du pH soit davantage lié à des modifications de la concentration en Ca2+ libre et en ATP [12].
Régulation purinergique
La boucle cytoplasmique des RyR2 présente un site de fixation pour l’ATP. En présence de Ca2+, l’ATP joue un rôle activateur. Cependant, tout comme celle du Mg2+, la concentration en ATP est relativement stable au cours du CEC normal. Le rôle de l’ATP semble à prendre en considération dans des conditions d’ischémie au cours desquelles sa concentration peut chuter et mener à une inhibition de l’activité des RyR2. Dans les cellules non excitables, le cADPR peut activer directement les RyR2. Cette activation n’a pas été retrouvée dans le myocarde. Le rôle du cADPR pourrait être indirect et passer par la production d’oxyde nitrique (NO).
Régulation par le potentiel redox
Chaque monomère de RyR2 contient 80 à 100 résidus cystéines dont 25 à 50 sont dans un état réduit, et 8 d’entre eux considérés comme hyper réactifs à des réactions d’oxydation [13]. De façon générale, les éléments réducteurs ont un effet inhibiteur sur le canal alors que les oxydants sont activateurs. Le glutathion, qui est le tampon redox prédominant dans les cellules eucaryotes, inhibe le canal dans sa forme réduite (GSH) et l’active dans sa forme oxydée (GSSG). L’oxyde nitrique est également considéré comme un modulateur physiologique de l’état redox du RyR2 par S-nitrosylation [13]. L’oxydation de certaines fonctions thiols modifie la réponse du canal à des agents modulateurs comme les nucléotides ou la caféine, et modifie la sensibilité du canal au Ca2+ et au Mg2+. Les effets d’agents oxydo-réducteurs peuvent donc être directs ou également modifier les interactions de type protéine-protéine du complexe macromoléculaire. Dans ce contexte, il a été montré que des agents oxydants peuvent dissocier la calmoduline [14] et, plus récemment, le calstabine 2 [15] du RyR2.
Pharmacologie des RyR2
Le récepteur de la ryanodine doit son nom à cet alcaloïde végétal qu’il fixe avec une très grande spécificité. La ryanodine se fixe sur deux sites de haute et de faible affinité situés sur l’extrémité carboxy-terminale [16] ; à faible concentration (~10 nM), elle maintient le canal dans un état de sous-conductance qui conduit à une fuite de Ca2+ et à la vidange du RS. A forte concentration (>1 µm), le canal est maintenu dans un état fermé. Le rouge de ruthénium est également un bloqueur puissant des RyR2, mais il est plus rarement utilisé en raison de sa faible spécificité pour les RyR2.
La caféine est un autre alcaloïde végétal très employé pour l’analyse fonctionnelle du RyR2 et du niveau de charge calcique du RS. Cet agent, en se fixant sur le RyR2, augmente la Po du canal, les temps moyens d’ouverture et induit une libération importante et transitoire de Ca2+ qui conduit à une vidange rapide du RS.
Les RyR2 sont très sensibles aux anesthésiques couramment utilisés en clinique. Les composés volatiles comme l’halothane ou l’isoflurane exercent un effet agoniste sur le canal. Les anesthésiques locaux (tétracaïne, procaïne, bupivacaïne) sont agonistes ou antagonistes selon la molécule utilisée.
Des efforts importants ont récemment porté sur la mise au point d’un dérivé benzothiazépine plus communément appelé JTV-519. L’analyse des effets de cette molécule indique qu’elle stabilise l’état fermé des RyR2 en diastole via une meilleure fixation de la calstabine 2 ; ce qui résulte, sur le plan fonctionnel, en une diminution de la fuite spontanée de Ca2+ du RS [17]. Par cette action sur le RyR2, cette molécule améliorerait la fonction systolique et présenterait un potentiel anti-arythmique important [17, 18].
La mise en évidence du site d’interaction du JTV-519 sur la séquence des RyR2 a récemment permis de proposer un modèle moléculaire du mode d’action de cette drogue [36].
Rôle des RyR2 dans le contrôle de la contraction et du rythme cardiaque
L’entrée de Ca2+ au cours du PA déclenche l’ouverture des RyR2 et la libération du Ca2+ du RS : c’est le Ca2+-induced Ca2+-release (CICR). L’ouverture d’un seul DHPR est suffisante pour déclencher une libération élémentaire de Ca2+ (Ca2+-spark) générée par un petit groupe de RyR2. La sommation de milliers de sparks associés au courant calcique de type L (ICa,L) permet une augmentation massive et transitoire du Ca2+ cytosolique déclenchant la contraction. L’amplitude de ces variations transitoires de [Ca2+]i est proportionnelle à ICa,L et à la charge du RS. La libération de Ca2+ du RS décrit une courbe en cloche en fonction de la concentration Ca2+. En effet, le RyR2 s’active pour des concentrations de Ca2+ <100 nM, et atteint un maximum à plus de 10 µM. La Po du canal devient très faible pour des concentrations de l’ordre du mM, et le canal s’inactive.
En plus de leur rôle essentiel pour la contraction cardiaque, les RyR2 modulent constamment le niveau de Ca2+ cytosolique (systolique et diastolique) qui est un facteur de régulation important de différents canaux et transporteurs ioniques membranaires. On peut citer le canal calcique de type L, les canaux chlorure activés par le Ca2+, le canal potassique à rectification entrante (IK1) et l’échangeur Na+/Ca2+. Via ces effets, le RyR2 devient un acteur majeur de la régulation du rythme cardiaque.
RyR2 et dysfonctionnement myocardique
Au vu des rôles majeurs joués par le RyR2 dans le fonctionnement normal des cellules cardiaques via différents modes de régulation et de la complexité de sa structure, il apparaît intuitivement qu’une dysfonction du RyR2 ou de l’une de ses protéines partenaires a des conséquences fonctionnelles importantes. La littérature abonde dans ce sens. Des dysfonctionnements des RyR2 sont associés à des troubles du CEC et de l’activité électrique. Récemment, on a découvert que certains dysfonctionnements liés à des mutations naturelles du RyR2 sont liés à des troubles du rythme sévères qui peuvent conduire à une mort subite ; c’est le cas, par exemple, des tachycardies ventriculaires polymorphes catécholaminergiques (TVPC) [19, 20]. Mais certaines pathologies acquises, comme l’insuffisance cardiaque, induisent également un dysfonctionnement des RyR2 qui semble impliquer des interactions protéine-protéine, comme dans le cas de la calstabine 2.
Tachycardies ventriculaires polymorphes catécholaminergiques
Les tachycardies ventriculaires polymorphes catécholaminergiques(TVPC) liées à des mutations des RyR2 représentent un trouble du rythme assez rare qui, contrairement aux syndromes du QT long, n’est pas observé chez le nourrisson [35]. Les TVCP se révèlent plus tardivement, typiquement lors de l’effort ou du stress et en l’absence de toute altération morphologique ou structurale du myocarde, avec un risque élevé de mort subite fatale chez l’adolescent(e) et le(a) jeune adulte. Les tachycardies ventriculaires associées à un QT long reflètent une anomalie d’un canal ionique (Na+, K+, Ca2+) qui entraîne une repolarisation anormale du potentiel d’action [21, 35]. Les TVPC sont liées principalement à une dérégulation des mouvements calciques intracellulaires, en particulier du fait d’une augmentation spontanée de l’activité des RyR2 générant une fuite de calcium y compris pendant la diastole. Le Ca2+ libéré de manière intempestive a pour conséquence de moduler l’activité de canaux ioniques Ca2+-dépendants qui deviennent alors sources d’arythmies. Ces effets sont exacerbés lors d’une stimulation β-adrénergique, principalement en stimulant le niveau de charge du réticulum sarcoplasmique et, donc, en augmentant la libération calcique via les RyR2 [22].
C’est au cours des années 2000 que le lien entre ces arythmies malignes, responsables de mort subite, et des mutations du gène codant pour le RyR2 (chromosome 1q42.1-43) a été découvert [19, 20]. À ce jour, 69 mutations ont été identifiées et recensées dans une revue très récente [19, 20]. Les mutations ont été identifiées au niveau de quatre régions du gène codant pour RyR2 : CPVT-I (77-466), CPVT-II (2246-2534), CPVT-III (3778-4201) et CPVT-IV (4497-4959) et semblent impliquer une altération des interactions entre domaines au sein du RyR2, des perturbations du complexe macromoléculaire et une modification de la sensibilité au Ca2+ cytoplasmique. Curieusement, aucune mutation au niveau du pore, essentiel à la perméation, et au niveau de la partie carboxy-terminale extrême du RyR2 impliquée dans la stabilité du canal, n’ont été rapportées [19, 20].
Sur le plan fonctionnel, plusieurs mécanismes résultant de ces mutations ont été évoqués comme une augmentation de l’affinité du canal pour le Ca2+ sur la face luminale du RS ou cytoplasmique, ou encore une dissociation de la protéine régulatrice FKBP12.6. Ces mutations sont responsables d’une activité anormale du canal et d’une altération de l’homéostasie calcique diastolique qui semble conduire essentiellement à des post-dépolarisations tardives au cours de stress catécholaminergiques. Les β-bloqueurs, bien que d’une efficacité aléatoire et relative, restent à ce jour un traitement couramment utilisé. Dans les situations où une dissociation de la protéine partenaire FKBP12.6 interviendrait, un traitement par le JTV-519, ou par l’un de ses analogues fonctionnels, semble une approche thérapeutique à considérer [23].
Des TVCP ont également été associées à des mutations portées par la calséquestrine qui peuvent conduire à des anomalies de fonctionnement des RyR2 par un mécanisme qui reste à déterminer [24].
Insuffisance cardiaque
Au cours de l’IC, l’homéostasie calcique est profondément altérée avec une diminution de la charge du RS et des variations transitoires de [Ca2+]i (diminution du calcium systolique). Ces modifications sont associées à une augmentation de [Ca2+]i au cours de la diastole. Ces détériorations ont été attribuées à un défaut de recaptage du Ca2+ vers le RS par la pompe SERCA ainsi qu’à un dysfonctionnement des RyR2. Il a ainsi été proposé qu’au cours de l’IC, la stimulation β-adrénergique chronique est à l’origine d’une hyperphosphorylation du canal conduisant à la dissociation de la calstabine 2 des RyR2 qui provoque : (1) en systole, la désynchronisation des ouvertures et fermetures du canal, avec un ralentissement des cinétiques ainsi qu’à la diminution de l’amplitude du signal calcique transitoire [25], et (2) en diastole à une augmentation de la fuite du Ca2+, contribuant ainsi à la diminution de la charge en Ca2+ du RS [26].
Les β-bloqueurs favoriseraient la stabilité du complexe calstabine 2 / RyR2 et normaliseraient l’activité du canal [27]. Cependant, au cours de l’IC, la stimulation sympathique chronique tend à faire diminuer la densité des récepteurs β-adrénergiques et plus particulièrement l’isoforme β1. L’analyse du complexe macromoléculaire au cours de l’IC démontre que cette contradiction résulte de la perte du complexe canalaire des phosphatases et de la PDE4D3 [28].
L’importance de l’interaction RyR2-calstabine 2 dans les processus physiopathologiques semble plutôt admise à ce jour. Cependant, le concept de la dissociation de la calstabine 2 induite par la phosphorylation du RyR2 sur la sérine 2808 au cours de l’IC est très controversé. L’utilisation récente d’une souris transgénique pour un récepteur RyR2 dans lequel la sérine 2808, phosphorylable par la PKA, a été substituée par une alanine non phosphorylable, semble indiquer une régulation β-adrénergique normale et exclure le rôle de la phosphorylation du RyR2 sur la sérine 2808 dans le développement de l’insuffisance cardiaque [29].
Ainsi, certains auteurs privilégient un dysfonctionnement des RyR2 médiée par la CamKII plutôt que par la PKA. Cela reste encore très largement débattu. Cependant, l’exploration physiopathologique du rôle d’EPAC (exchange protein directly activated by cAMP), voie nouvellement caractérisée dans la régulation des RyR2 [30, 31], permettra peut être de mieux appréhender les divergences des résultats observés.
RyR2 et arythmies
De nombreux décès de patients porteurs d’une cardiopathie ischémique ou d’une hypertrophie cardiaque, comme chez ceux atteints de TVCP, sont dus à des troubles du rythme ventriculaires (résultant de tachycardies ventriculaires, de fibrillations ventriculaires et de blocs auriculo-ventriculaire). Au niveau cellulaire, les post-dépolarisations tardives (DAD, delayed after depolarisation) ou précoces (EAD, early after depolarisation) sont les deux troubles majeurs de la repolarisation. Ils sont généralement associés à des modifications abruptes de la fréquence cardiaque. Les EAD (fibrillation ventriculaire) apparaissent essentiellement au cours d’une bradycardie alors que les DAD sont associées à une augmentation de la fréquence cardiaque [32]. Comme cela est décrit précédemment, le rôle central des RyR2 au cours de changements de la fréquence cardiaque en fait un acteur essentiel des troubles du rythme et de la mort subite, en particulier pendant la phase diastolique au cours de laquelle la fuite calcique peut déclencher des DAD.
La fréquence cardiaque est régulée par la stimulation β-adrénergique. Elle augmente le recaptage du Ca2+ dans le RS et, donc, augmente la fuite spontanée via les RyR2. Cette régulation physiologique normale peut s’avérer délétère chez des sujets dont les RyR2 présentent une dysfonction acquise ou chronique, et amplifier cette dysfonction jusqu’à l’établissement de troubles du rythme graves. En effet, l’augmentation de la [Ca2+]i qui en résulte augmente l’activité de l’échangeur Na+/Ca2+ au cours de la diastole favorisant ainsi la dépolarisation cellulaire. Nous avons récemment démontré que la fuite de Ca2+ diastolique, générée par les RyR2, avait pour conséquence de bloquer le courant repolarisant IK1 permettant à la cellule, en synergie avec l’échangeur Na+/Ca2+, de se dépolariser jusqu’au seuil de déclenchement d’un nouveau PA [33, 34]. La cellule entre alors dans un cycle auto-entretenu responsable de DAD [32] (Figure 2). Dans ce contexte, une approche pharmacologique ciblant spécifiquement le RyR2 en normalisant sa fonction peut s’avérer essentielle dans le traitement des troubles du rythme cardiaque diastoliques.
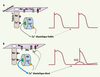 |
Figure 2. Importance de la [Ca2+]i de repos (c’est-à-dire diastolique) sur le contrôle du potentiel membranaire de cardiomyocytes ventriculaires. En situation normale (A), l’activité des RyR2 est insignifiante et la faible quantité de Ca2+ libérée pendant la diastole est rapidement repompée par le RS. Le potentiel de repos diastolique est essentiellement contrôlé et maintenu à une valeur de l’ordre de -80 mV par l’activité des canaux potassiques à rectification entrante (IK1). En cas d’anomalie de fonctionnement des RyR2 (B), on observe une fuite de calcium du RS pendant la diastole et une surchage cytosolique qui active le courant entrant dépolarisant de l’échangeur Na+/Ca2+ et inhibe le courant IK1. Cela se traduit par des dépolarisations membranaires spontanées pouvant atteindre le seuil d’excitation des canaux sodiques, et le déclenchement d’un potentiel d’action précoce ; on parle alors de post-dépolarisation tardive (DAD). |
Références
- Fill M, Copello JA. Ryanodine receptor calcium release channels. Physiol Rev 2002; 82 : 893–922. [Google Scholar]
- Bers DM. Macromolecular complexes regulating cardiac ryanodine receptor function. J Mol Cell Cardiol 2004; 37 : 417–29. [Google Scholar]
- Brillantes AB, Ondrias K, Scott A, et al. Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell 1994; 77 : 513–23. [Google Scholar]
- Balshaw DM, Xu L, Yamaguchi N, et al. Calmodulin binding and inhibition of cardiac muscle calcium release channel (ryanodine receptor). J Biol Chem 2001; 276 : 20144–53. [Google Scholar]
- Yamaguchi N, Takahashi N, Xu L, et al. Early cardiac hypertrophy in mice with impaired calmodulin regulation of cardiac muscle Ca release channel. J Clin Invest 2007; 117 : 1344–53. [Google Scholar]
- Kapiloff MS, Jackson N, Airhart N. mAKAP and the ryanodine receptor are part of a multi-component signaling complex on the cardiomyocyte nuclear envelope. J Cell Sci 2001; 114 : 3167–76. [Google Scholar]
- Witcher DR, Kovacs RJ, Schulman H, et al. Unique phosphorylation site on the cardiac ryanodine receptor regulates calcium channel activity. J Biol Chem 1991; 266 : 11144–52. [Google Scholar]
- Wehrens XH, Lehnart SE, Reiken SR, Marks AR. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ Res 2004; 94 : e61–70. [Google Scholar]
- Farrell EF, Antaramian A, Rueda A, et al. Sorcin inhibits calcium release and modulates excitation-contraction coupling in the heart. J Biol Chem 2003; 278 : 34660–6. [Google Scholar]
- Seidler T, Miller SL, Loughrey CM, et al. Effects of adenovirus-mediated sorcin overexpression on excitation-contraction coupling in isolated rabbit cardiomyocytes. Circ Res 2003; 93 : 132–9. [Google Scholar]
- Terentyev D, Viatchenko-Karpinski S, Valdivia HH, et al. Luminal Ca2+ controls termination and refractory behavior of Ca2+-induced Ca2+ release in cardiac myocytes. Circ Res 2002; 91 : 414–20. [Google Scholar]
- Xu L, Mann G, Meissner G. Regulation of cardiac Ca2+ release channel (ryanodine receptor) by Ca2+, H+, Mg2+, and adenine nucleotides under normal and simulated ischemic conditions. Circ Res 1996; 79 : 1100–9. [Google Scholar]
- Xu L, Eu JP, Meissner G, Stamler JS. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science 1998; 279 : 234–7. [Google Scholar]
- Zhang JZ, Wu Y, Williams BY, et al. Oxidation of the skeletal muscle Ca2+ release channel alters calmodulin binding. Am J Physiol 1999; 276 : C46–53. [Google Scholar]
- Zissimopoulos S, Docrat N, Lai FA. Redox sensitivity of the ryanodine receptor interaction with FK506-binding protein. J Biol Chem 2007; 282 : 6976–83. [Google Scholar]
- Callaway C, Seryshev A, Wang JP, et al. Localization of the high and low affinity 3Hryanodine binding sites on the skeletal muscle Ca2+ release channel. J Biol Chem 1994; 269 : 15876–84. [Google Scholar]
- Yano M, Kobayashi S, Kohno M, et al. FKBP12.6-mediated stabilization of calcium-release channel (ryanodine receptor) as a novel therapeutic strategy against heart failure. Circulation 2003; 107 : 477–84. [Google Scholar]
- Lehnart SE, Terrenoire C, Reiken S, et al. Stabilization of cardiac ryanodine receptor prevents intracellular calcium leak and arrhythmias. Proc Natl Acad Sci USA 2006; 103 : 7906–10. [Google Scholar]
- George CH, Jundi H, Thomas NL, et al. Ryanodine receptors and ventricular arrhythmias : emerging trends in mutations, mechanisms and therapies. J Mol Cell Cardiol 2007; 42 : 34–50. [Google Scholar]
- Priori SG, Napolitano C. Cardiac and skeletal muscle disorders caused by mutations in the intracellular Ca2+ release channels. J Clin Invest 2005; 115 : 2033–8. [Google Scholar]
- Pourrier M, Nattel S. Protéines d’ancrage et mort subite cardiaque : comment et pourquoi ? Med Sci (Paris) 2004; 20 : 437–41. [Google Scholar]
- Fernandez-Velasco M, Gomez AM, Richard S. Unzipping RyR2 in adult cardiomyocytes : getting closer to mechanisms of inherited ventricular arrhythmias ? Cardiovasc Res 2006; 70 : 407–9. [Google Scholar]
- Lehnart SE, Wehrens XH, Laitinen PJ, et al. Sudden death in familial polymorphic ventricular tachycardia associated with calcium release channel (ryanodine receptor) leak. Circulation 2004; 109 : 3208–14. [Google Scholar]
- Terentyev D, Nori A, Santoro M, et al. Abnormal interactions of calsequestrin with the ryanodine receptor calcium release channel complex linked to exercise-induced sudden cardiac death. Circ Res 2006; 98 : 1151–8. [Google Scholar]
- Litwin SE, Zhang D, Bridge JH. Dyssynchronous Ca2+ sparks in myocytes from infarcted hearts. Circ Res 2000; 87 : 1040–7. [Google Scholar]
- Marx SO, Reiken S, Hisamatsu Y, et al. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor) : defective regulation in failing hearts. Cell 2000; 101 : 365–76. [Google Scholar]
- Doi M, Yano M, Kobayashi S, et al. Propranolol prevents the development of heart failure by restoring FKBP12.6-mediated stabilization of ryanodine receptor. Circulation 2002; 105 : 1374–9. [Google Scholar]
- Lehnart SE, Wehrens XH, Reiken S, et al. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell 2005; 123 : 25–35. [Google Scholar]
- Benkusky NA, Weber CS, Scherman JA, et al. Intact beta-adrenergic response and unmodified progression toward heart failure in mice with genetic ablation of a major protein kinase A phosphorylation site in the cardiac ryanodine receptor. Circ Res 2007; 101 : 819–29. [Google Scholar]
- Oestreich EA, Wang H, Malik S, et al. Epac-mediated activation of phospholipase C(epsilon) plays a critical role in beta-adrenergic receptor-dependent enhancement of Ca2+ mobilization in cardiac myocytes. J Biol Chem 2007; 282 : 5488–95. [Google Scholar]
- Pereira L, Metrich M, Fernandez-Velasco M, et al. The cAMP binding protein Epac modulates Ca2+ sparks by a Ca2+/calmodulin kinase signalling pathway in rat cardiac myocytes. J Physiol 2007; 583 : 685–94. [Google Scholar]
- Bers DM. Calcium and cardiac rhythms : physiological and pathophysiological. Circ Res 2002; 90 : 14–7. [Google Scholar]
- Fauconnier J, Lacampagne A, Rauzier JM, et al. Frequency-dependent and proarrhythmogenic effects of FK-506 in rat ventricular cells. Am J Physiol Heart Circ Physiol 2005; 288 : H778–86. [Google Scholar]
- Fauconnier J, Lacampagne A, Rauzier JM, et al. Ca2+-dependent reduction of IK1 in rat ventricular cells : a novel paradigm for arrhythmia in heart failure ? Cardiovasc Res 2005; 68 : 204–12. [Google Scholar]
- Escande D. Aspects moléculaires de la mort subite de l’adulte. Med Sci (Paris) 2004; 20 : 623–5. [Google Scholar]
- Yamamoto T, Yano M, Xu X, et al. Identification of target domains of the cardiac ryanodine receptor to correct channel disorder in failing hearts. Circulation 2008; 117 : 762–72 [Google Scholar]
Liste des tableaux
Liste des principales protéines régulatrices associées au récepteur de la ryanodine et formant le complexe macromoléculaire.
Liste des figures
|
Figure 1. Représentation schématique du complexe macromoléculaire formé par le récepteur de la ryanodine et ses protéines associées. CaM : calmoduline ; CSQ : calsequestrine ; FKBP : calstabine 2. |
| Dans le texte | |
|
Figure 2. Importance de la [Ca2+]i de repos (c’est-à-dire diastolique) sur le contrôle du potentiel membranaire de cardiomyocytes ventriculaires. En situation normale (A), l’activité des RyR2 est insignifiante et la faible quantité de Ca2+ libérée pendant la diastole est rapidement repompée par le RS. Le potentiel de repos diastolique est essentiellement contrôlé et maintenu à une valeur de l’ordre de -80 mV par l’activité des canaux potassiques à rectification entrante (IK1). En cas d’anomalie de fonctionnement des RyR2 (B), on observe une fuite de calcium du RS pendant la diastole et une surchage cytosolique qui active le courant entrant dépolarisant de l’échangeur Na+/Ca2+ et inhibe le courant IK1. Cela se traduit par des dépolarisations membranaires spontanées pouvant atteindre le seuil d’excitation des canaux sodiques, et le déclenchement d’un potentiel d’action précoce ; on parle alors de post-dépolarisation tardive (DAD). |
| Dans le texte | |
Les statistiques affichées correspondent au cumul d'une part des vues des résumés de l'article et d'autre part des vues et téléchargements de l'article plein-texte (PDF, Full-HTML, ePub... selon les formats disponibles) sur la platefome Vision4Press.
Les statistiques sont disponibles avec un délai de 48 à 96 heures et sont mises à jour quotidiennement en semaine.
Le chargement des statistiques peut être long.