")
")
| Issue |
Med Sci (Paris)
Volume 24, Number 4, Avril 2008
|
|
|---|---|---|
| Page(s) | 341 - 343 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/2008244341 | |
| Published online | 15 avril 2008 | |
L’augmentation du HDL-cholestérol
Un traitement d’avenir dans le domaine de l’athérosclérose ?
Raising HDL-cholesterol: the future drug therapy for atherosclerosis ?
Division of Molecular Medicine, Department of Medicine, Columbia University, 630 West 168 th Street, New York, NY 10032, États-Unis
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
L’athérosclérose est la cause majeure des maladies cardiovasculaires et est associée à environ 50 % des décès dans les pays industrialisés. Les études épidémiologiques révèlent depuis longtemps une association inverse entre les niveaux de HDL (high density lipoproteins)-cholestérol plasmatique et les maladies cardiovasculaires [1]. Des médicaments visant à augmenter ce « bon » cholestérol existent déjà en thérapeutique humaine, mais des avancées récentes dans la compréhension du rôle protecteur des HDL, associées au développement de nouvelles molécules, pourraient entraîner une généralisation rapide de cette classe thérapeutique dans le traitement de l’athérosclérose [2].
Le transport inverse du cholestérol
L’athérosclérose est caractérisée par la formation de la plaque d’athérome avec accumulation de lipides, cellules apoptotiques et éléments fibreux constituant des sites inflammatoires dans la paroi artérielle [3]. Classiquement, on considère que les effets protecteurs des HDL et de leur apoliprotéine majeure, l’apoA-1, sont liés à leur rôle d’épuration du cholestérol des macrophages spumeux présents dans la plaque [4]. Ce mécanisme représente la première étape du processus de transport inverse du cholestérol, voie métabolique impliquant le retour du cholestérol vers le foie où il pourra être éliminé dans la bile. Dans la circulation, les HDL peuvent être remodelées pour faciliter ce retour grâce à différentes enzymes dont la LCAT (lecithin cholesterol acyltransferase) permettant l’estérification du cholestérol, et la CETP (cholesterol ester transfer protein) facilitant le transfert d’esters de cholestérol des HDL vers les VLDL (very low density lipoproteins) et LDL (low density lipoproteins) (Figure 1).
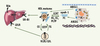 |
Figure 1. Le transport inverse du cholestérol. L’apolipoprotéine A-1 (apoA-1) interagit avec les transporteurs ABCA1 présents dans les macrophages de la plaque athéromateuse et induisent l’efflux de cholestérol entraînant ainsi la formation de HDL. Sous l’action de la LCAT (lecithin cholesterol acyltransferase), le cholestérol libre (FC, free cholesterol) sera estérifié en esters de cholestérol (CE, cholesteryl esters) permettant la maturation des HDL. Les HDL matures deviendront alors les accepteurs de l’efflux de cholestérol induit entre autres par ABCG1. Un relargage de l’apoA-I des HDL peut s’effectuer sous l’action de différentes enzymes tels que des lipases (HL, hepatic lipase et EL, endothelial lipase) ou la PLTP (phospholipid transfer protein). Chez l’homme, les esters de cholestérol peuvent être transférés des HDL matures vers les VLDL et LDL sous l’action de la CETP (cholesterol ester transfer protein) favorisant l’élimination du cholestérol par la voie du LDLR hépatique (low-density lipoprotein receptor). Le cholestérol des HDL matures quant à lui sera éliminé par le foie dans la bile après captation par la voie du SR-BI (scavenger receptor class BI). |
Les ABC transporteurs et l’épuration du cholestérol de la plaque athéromateuse
Deux transporteurs de la famille des ATP-binding cassette (ABC) ont été identifiés comme permettant l’efflux de cholestérol des macrophages vers les HDL. Le transporteur ABCA1, molécule mutante dans la maladie de Tangier, assure l’efflux de cholestérol et de phospholipides vers l’apoliprotéine A-1 non lipidée participant ainsi à la formation des HDL [5]. Les transporteurs ABCG1 quant à eux, permettent l’efflux de cholestérol spécifiquement vers les HDL matures [6]. Ainsi, ces 2 transporteurs agiraient de manière synergique pour éviter l’accumulation de cholestérol dans les macrophages (Figure 2). Leur rôle central a récemment été mis en évidence dans une étude de transplantation de moelle osseuse de souris donneuses déficientes pour ABCA1 et ABCG1 chez des souris hyperlipidémiques. Ces souris présentent un phénotype remarquable caractérisé par l’accumulation de cellules spumeuses dans de nombreux tissus et par une susceptibilité très forte à l’athérosclérose [7].
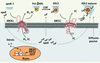 |
Figure 2. Les mécanismes d’efflux du cholestérol. Les mécanismes d’efflux du cholestérol sont gouvernés par le contenu intracellulaire en stérols, ligands naturels des récepteurs nucléaires LXR (liver X receptor). Deux transporteurs de la famille des ATP-binding cassette (ABCA1 et ABCG1) ainsi que l’apolipoprotéine E (apoE) ont été identifiés comme des gènes cibles de ces facteurs de transcription. Les transporteurs ABCA1 et ABCG1 agiraient de manière synergique pour assurer successivement l’efflux de cholestérol libre (FC) et de phospholipides (PL) vers l’apoA-1 non lipidée et les HDL matures. L’enzyme LCAT (lecithin cholesterol acyltransferase) faciliterait l’efflux en estérifiant le cholestérol libre en esters de cholestérol (CE), générant ainsi un gradient de cholestérol libre en faveur de l’efflux. L’apoE aurait un rôle à toutes les étapes de ce processus en interagissant avec les transporteurs ABCA1, en stimulant l’activité LCAT, et en améliorant la capacité des HDL à induire l’efflux de cholestérol par les transporteurs ABCG1. La diffusion passive participerait également aux mécanismes d’efflux du cholestérol non contrôlés par le facteur de transcription LXR. |
L’apoE et l’efflux de cholestérol
L’apoE pourrait avoir un rôle majeur dans le transport du cholestérol lié aux transporteurs ABCA1 et ABCG1. En effet, l’apoE a la capacité d’interagir avec ABCA1 et l’accumulation d’apoE dans les HDL leur conférerait une meilleure capacité à induire l’efflux de cholestérol par les transporteurs ABCG1 [5, 7, 8]. De plus, contrairement à l’apoA-1, l’apoE est sécrétée au sein de la plaque athéromateuse par les macrophages présents et pourrait ainsi avoir un rôle local (Figure 2).
Les HDL et l’inflammation des macrophages dans la plaque athéromateuse
L’athérosclérose est caractérisée par une inflammation qui est initiée par la rétention des lipoprotéines LDL dans la paroi artérielle [9]. Les LDL piégées subissent des modifications oxydatives menant à une cascade d’événements comprenant la réponse inflammatoire des macrophages. Les HDL auraient un effet bénéfique à toutes les étapes de ce processus inflammatoire [10]. Les HDL, par compétition, peuvent prévenir la rétention des LDL et auraient également un pouvoir anti-oxydant permettant de limiter leur oxydation. Une fois oxydées, les LDL sont capturées par les macrophages infiltrés entraînant une accumulation de lipides associée à la sécrétion d’un panel de facteurs inflammatoires caractéristiques des cellules spumeuses et conduisant finalement à l’apoptose des cellules [9, 10]. Les HDL, en induisant l’efflux de cholestérol par l’intermédiaire d’ABCA1 et ABCG1, auraient donc des effets anti-inflammatoires et anti-apoptotiques, ce qui limiterait le risque d’une rupture de la plaque et la formation d’un thrombus occluant la lumière du vaisseau sanguin [7].
L’augmentation du bon HDL-cholestérol dans le traitement de l’athérosclérose, une perspective thérapeutique
Parallèlement aux statines qui agissent principalement en diminuant le « mauvais » LDL-cholestérol, une place de choix pourrait donc être donnée aux molécules ciblant le HDL-cholestérol dans le traitement de l’athérosclérose [2, 11]. Parmi les molécules actuellement utilisées ou en cours de développement, on peut distinguer les médicaments augmentant les niveaux de HDL-cholestérol dans la circulation, comme les inhibiteurs CETP, la niacine, les agonistes des récepteurs nucléaires PPAR. Il est important de noter que malgré l’échec du Torcetrapib, le développement des inhibiteurs CETP n’est pas abandonné puisqu’il apparaît clairement que cette molécule présentait des effets délétères indépendants de son action inhibitrice de la CETP. L’élucidation de nouveaux mécanismes de régulation du transport inverse du cholestérol pourrait permettre, cependant, l’élaboration de nouvelles approches thérapeutiques visant a augmenter les niveaux plasmatiques de HDL-cholestérol. Dans une deuxième catégorie, on peut citer l’administration de particules permettant l’efflux de cholestérol (apoA-1 mimétiques), ou les agonistes du récepteur nucléaire LXR (liver X receptor) agissant directement sur la paroi vasculaire en stimulant l’efflux de cholestérol via les transporteurs ABCA1 et ABCG1 (Figure 2). L’avenir devrait très prochainement nous dire si ces traitements sont efficaces pour lutter contre les maladies cardiovasculaires et prolonger ainsi l’espérance de vie.
Remerciements
Les auteurs remercient le Pr Alan R. Tall pour la discussion scientifique.
Références
- Gordon DJ, Rifkind BM. High-density lipoprotein-the clinical implications of recent studies. N Engl J Med 1989; 321 : 1311–6. [Google Scholar]
- Linsel-Nitschke P, Tall AR. HDL as a target in the treatment of atherosclerotic cardiovascular disease. Nat Rev 2005; 4 : 193–205. [Google Scholar]
- Ross, R. Atherosclerosis-an inflammatory disease. N Engl J Med 1999; 340 : 115–26. [Google Scholar]
- Cuchel M, Rader DJ. Macrophage reverse cholesterol transport: key to the regression of atherosclerosis. Circulation 2006; 113 : 2548–55. [Google Scholar]
- Oram JF, Lawn RM, Garvin MR, Wade DP. ABCA1 is the cAMP-inducible apolipoprotein receptor that mediates cholesterol secretion from macrophages. J Biol Chem 2000; 275 : 34508–11. [Google Scholar]
- Wang N, Lan D, Chen W, et al. ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Proc Natl Acad Sci USA 2004; 101 : 9774–9. [Google Scholar]
- Yvan-Charvet L, Ranalletta M, Wang N, et al. Combined deficiency of ABCA1 and ABCG1 promotes foam cell accumulation and accelerates atherosclerosis in mice. J Clin Invest 2007; 117 : 3900–8. [Google Scholar]
- Matsuura F, Wang N, Chen W, et al. HDL from CETP-deficient subjects shows enhanced ability to promote cholesterol efflux from macrophages in an apoE- and ABCG1-dependent pathway. J Clin Invest 2006; 116 : 1435–42. [Google Scholar]
- Aikawa M, Libby P. The vulnerable atherosclerotic plaque: pathogenesis and therapeutic approach. Cardiovasc Pathol 2004; 13 : 125–38. [Google Scholar]
- Kontush A, Chapman MJ. Functionally defective high-density lipoprotein: a new therapeutic target at the crossroads of dyslipidemia, inflammation, and atherosclerosis. Pharmacol Rev 2006; 58 : 342–74. [Google Scholar]
- Morozova S, Suc-Royer I, Auwerx J. Modulateurs du métabolisme du cholestérol et avenir du traitement de l’athérosclérose. Med Sci (Paris) 2004; 20 : 685–90. [Google Scholar]
© 2008 médecine/sciences - Inserm / SRMS
Liste des figures
|
Figure 1. Le transport inverse du cholestérol. L’apolipoprotéine A-1 (apoA-1) interagit avec les transporteurs ABCA1 présents dans les macrophages de la plaque athéromateuse et induisent l’efflux de cholestérol entraînant ainsi la formation de HDL. Sous l’action de la LCAT (lecithin cholesterol acyltransferase), le cholestérol libre (FC, free cholesterol) sera estérifié en esters de cholestérol (CE, cholesteryl esters) permettant la maturation des HDL. Les HDL matures deviendront alors les accepteurs de l’efflux de cholestérol induit entre autres par ABCG1. Un relargage de l’apoA-I des HDL peut s’effectuer sous l’action de différentes enzymes tels que des lipases (HL, hepatic lipase et EL, endothelial lipase) ou la PLTP (phospholipid transfer protein). Chez l’homme, les esters de cholestérol peuvent être transférés des HDL matures vers les VLDL et LDL sous l’action de la CETP (cholesterol ester transfer protein) favorisant l’élimination du cholestérol par la voie du LDLR hépatique (low-density lipoprotein receptor). Le cholestérol des HDL matures quant à lui sera éliminé par le foie dans la bile après captation par la voie du SR-BI (scavenger receptor class BI). |
| Dans le texte | |
|
Figure 2. Les mécanismes d’efflux du cholestérol. Les mécanismes d’efflux du cholestérol sont gouvernés par le contenu intracellulaire en stérols, ligands naturels des récepteurs nucléaires LXR (liver X receptor). Deux transporteurs de la famille des ATP-binding cassette (ABCA1 et ABCG1) ainsi que l’apolipoprotéine E (apoE) ont été identifiés comme des gènes cibles de ces facteurs de transcription. Les transporteurs ABCA1 et ABCG1 agiraient de manière synergique pour assurer successivement l’efflux de cholestérol libre (FC) et de phospholipides (PL) vers l’apoA-1 non lipidée et les HDL matures. L’enzyme LCAT (lecithin cholesterol acyltransferase) faciliterait l’efflux en estérifiant le cholestérol libre en esters de cholestérol (CE), générant ainsi un gradient de cholestérol libre en faveur de l’efflux. L’apoE aurait un rôle à toutes les étapes de ce processus en interagissant avec les transporteurs ABCA1, en stimulant l’activité LCAT, et en améliorant la capacité des HDL à induire l’efflux de cholestérol par les transporteurs ABCG1. La diffusion passive participerait également aux mécanismes d’efflux du cholestérol non contrôlés par le facteur de transcription LXR. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.