")
")
| Issue |
Med Sci (Paris)
Volume 24, Number 3, Mars 2008
|
|
|---|---|---|
| Page(s) | 290 - 296 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/2008243290 | |
| Published online | 15 mars 2008 | |
Contribution De La Régulation post-transcriptionnelle À L’émergence De Maladies
L’enfance De L’are
Aberrant regulation of mRNA 3’ untranslated region in cancers and inflammation
Université de Toulouse, CBD, UMR, 5547, CNRS, IFR 109, 118, route de Narbonne, 31062 Toulouse, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
De nombreux ARN messagers (ARNm) portent dans leur région 3’ non codante une séquence riche en adénosines et uridines (ARE) qui constitue un élément régulateur clé de leur expression. L’ARE dicte la durée de vie et/ou la traductibilité de l’ARN qui le porte, via son interaction avec des protéines spécifiques, les AUBP, et des miARN. Il permet ainsi à la cellule de s’adapter à un changement environnemental en modulant de manière coordonnée, rapide et transitoire, le niveau d’expression d’un certain nombre d’ARNm. Étant donné que la plupart des protéines impliquées dans la réponse au stress et les réponses immune et inflammatoire, la croissance et la prolifération cellulaire sont codées par des ARNm portant un ARE, toute altération de l’ARE ou de l’expression des AUBP peut provoquer une synthèse protéique anormale à l’origine du développement de maladies.
Abstract
Almost 10 % of mammalian coding mRNAs contain in their 3’ untranslated region a sequence rich in adenine and uridine residues known as AU-rich element (ARE). Many of them encode oncogenes (for instance c-Myc and c-Fos), cell cycle regulators (cyclin D1, A1, B1), cytokines (TNFα, IL2) and growth factors (GM-CSF) which are overexpressed in cancer or inflammatory diseases due to increased mRNA stability and/or translation. AREs are recognized by a group of proteins, collectively called AUBPs which display various functions. For instance, HuR/ELAV is mainly known to protect ARE-containing mRNAs from degradation, while AUF1, TTP and KSRP act to destabilize their bound target mRNAs and TIA/TIAR to inhibit their translation. Alterations in ARE sequences or AUBP abundance, cellular localization or activity due to post-translational modifications such as phosphorylation can promote or enhance malignancy or perturb immune homeostasis. Here, c-myc and TNFα are chosen as examples to illustrate how altered 3’ UTR gene regulation impacts on pathologies.
© 2008 médecine/sciences - Inserm / SRMS
Le décor : ARE et AUBP
Les ARNm n’ont pas tous la même durée de vie, certains sont très stables (plusieurs jours), d’autres instables (moins d’une heure). La durée de vie de l’ARNm conditionne la fenêtre de temps durant laquelle il peut être traduit et détermine ainsi le niveau d’expression du gène correspondant [1, 2]. Le motif d’instabilité le plus étudié dans les cellules de mammifères est l’ARE, une séquence riche en adénines et uridines présente dans la région 3’ non codante (3’ UTR) de l’ARN. Découvert il y a vingt ans par Shaw et Kamen dans l’ARN d’un facteur de croissance, le GM-CSF (granulocyte macrophage colony-stimulating factor) [3], cet élément a depuis été identifié par analyse bioinformatique dans environ 8 % des ARNm codants [4]. Plusieurs classes d’ARE ont été décrites, selon que l’ARE contient ou non le pentamère AUUUA au sein d’une séquence riche en adénines et uridines (Figure 1). L’absence ou la présence du pentamère AUUUA, isolé ou réitéré, chevauchant ou non, a permis une première classification des ARE en trois catégories [5]. Ultérieurement, l’analyse bioinformatique des régions 3’UTR, fondée sur la recherche du motif WWWU(AUUUA)UWWW (W = A ou U), a permis de subdiviser la catégorie II, contenant 2 pentamères ou plus, en 4 groupes selon le nombre de pentamères présents [4]. L’ARE, un élément conservé chez la levure [6], n’agit pas tout seul mais en interaction avec des protéines, les AUBP (AU binding proteins) [7–9]. On trouve les orthologues de ces protéines chez la drosophile [10], ce qui suggère une conservation de la fonction régulatrice des ARE à travers l’évolution.
Principalement nucléaires, les AUBP font néanmoins la navette entre le noyau et le cytoplasme et participent ainsi à différents aspects de la vie d’un ARN à ARE : transport nucléo-cytoplasmique, contrôle de sa stabilité ou de sa traduction dans le cytoplasme. Certaines AUBP, comme celles de la famille ELAV/Hu, préservent l’ARN à ARE de la dégradation ; d’autres, telles que KSRP, TTP (tristetraprolin) ou les protéines apparentées BRF (butyrate response factor), provoquent sa dégradation. D’autres, comme TIA-1 ou TIAR, ne semblent pas intervenir directement dans la (dé)stabilisation des ARN à ARE mais régulent leur traductibilité. Enfin, certaines comme AUF1/hnRNPD ont un double, voire même un triple jeu, car elles accélèrent ou inhibent la dégradation et contrôlent la traduction de certains ARN [11]. Toutes ces protéines peuvent, en théorie, se fixer à une même séquence ARE et ainsi agir en synergie ou en compétition les unes avec les autres [11, 12] (Figure 1).
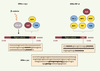 |
Figure 1. Diversité des séquences ARE et des AUBP avec lesquelles elles interagissent. La classification initiale des ARE en trois catégories [5] s’est enrichie de subdivisions de la catégorie II en 4 groupes suivant le nombre de pentamères AUUUA [4]. L’ARE de l’ARN c-myc (nt 2061-2141 NM_002467) illustre la catégorie I et celle du TNFa la catégorie II, groupe 3 (nt n° 1311-1385 du gène TNFα NM_000594). À titre de comparaison, l’ARE de c-jun, un exemple de la catégorie III qui ne contient pas de pentamère mais une région riche en uridines ainsi que plusieurs motifs GUUUG est indiqué (nt 2401-2542 du gène c-jun J04115). Deux AUUUA dispersés dans un contexte riche en uridines sont présents dans l’ARE de c-myc, alors que le pentamère se répète 8 fois, dont 6 de manière chevauchante, pour l’ARE du TNFa. Les AUBP interagissant avec ces 2 dernières séquences sont indiquées (en jaune les AUBP déstabilisantes, en rouge foncé la seule AUBP stabilisante et en bleu les régulateurs traductionnels). miR16 contient la séquence UAAAUAUU et peut donc s’apparier à plusieurs sites de l’ARE du TNFα [20]. La région codée par l’exon 3 de c-myc contient la séquence CRD reconnue par la protéine CRD-BP qui la protège d’action d’endonucléases (pour revue, voir [41]) et dont l’expression est activée par la voie β-caténine/Tcf. |
Les règles du jeu
Les AUBP peuvent être considérées comme des protéines adaptatrices ne possédant pas en elles-mêmes de fonction enzymatique particulière mais interagissant avec les machineries cellulaires intervenant dans la dégradation, la traduction, le stockage sélectif et la surveillance des ARNm [13]. L’exosome, un ensemble d’exoribonucléases à activité 3’ → 5’, a longtemps été considéré comme le seul acteur de la dégradation des ARN à ARE [14]. De petits granules cytoplasmiques concentrant des ARNm à ARE, une déadénylase et des composants de l’exosome viennent d’ailleurs d’être décrits [15]. Cependant les ARNm à ARE sont également présents dans d’autres foyers cytoplasmiques, les processing bodies (P bodies), caractérisés par des enzymes qui coupent la coiffe en 5’ de l’ARNm (DCP1/DCP2), l’exoribonucléase 5’ → 3’ XRN1 et également des composants clefs de la machinerie de dégradation associée aux microARN, le complexe RISC (RNA induced silencing complex) (revue dans [16, 17]). Ces P bodies partagent un certain nombre d’acteurs avec les granules de stress (SG), d’autres structures cytoplasmiques qui apparaissent, comme leur nom l’indique, en condition de stress [18]. Par exemple, TTP, une AUBP déstabilisante, favoriserait transitoirement leur association physique. Ainsi, en fonction de l’état de la cellule, les ARNm à ARE pourraient être stockés dans différents compartiments cellulaires, dégradés par l’une ou l’autre de leurs extrémités ou dirigés, après « réhabillage », vers les polysomes pour y être traduits (Figure 2). Une belle illustration de cette hypothèse est donnée par l’étude du gène CAT1 (cationic amino acid transporter 1) dans une lignée cellulaire d’hépatome humain. Lorsque ces cellules subissent un stress (par exemple la privation en acides aminés), la synthèse de CAT1 est considérablement accrue, essentiellement du fait d’une augmentation de la traduction de l’ARNm CAT1. Sans stress, cet ARNm à ARE est normalement adressé aux P bodies grâce à son association à un petit ARN non codant, miR122, complémentaire de sa région 3’ UTR. Mais lorsque la cellule est stressée, l’ARNm CAT1 est exclu des P bodies et activement traduit [19] (Figure 2). Ce changement pourrait être expliqué par une relocalisation, en réponse au stress, de l’AUBP stabilisante HuR du noyau vers le cytoplasme. Cette étude renforce l’hypothèse, initialement émise par Jing et al., de l’implication conjointe d’AUBP et de miARN dans le contrôle du métabolisme des ARNm à ARE [20]. Dans leur travail réalisé sur des cellules de drosophile, les auteurs montraient ainsi que TTP, en interagissant avec des protéines clefs du système miARN/ARNi, recrute un petit ARN, miR16, complémentaire de la séquence ARE du TNFα (tumour necrosis factor), permettant ainsi la dégradation de l’ARNm rapporteur contenant cet ARE.
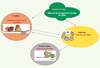 |
Figure 2. Localisation des ARNm dans divers granules cytoplasmiques. Les ARNm sont localisés dans différentes structures cytoplasmiques où ils sont stockés (granules de stress et P bodies) ou dégradés (exosomes bodies et P bodies). En fonction du stimulus cellulaire, les ARNm associés à différentes protéines peuvent être libérés de ces granules et activement traduits dans les polysomes (d’après [16–19]). |
Les transgressions
Quelles que soient les incertitudes sur les lieux et le « manège » des acteurs de la dégradation, on imagine bien que l’altération des ARE ou un changement dans le niveau d’expression des AUBP, de leur localisation sub-cellulaire ou de leur capacité effective à fixer l’ARE, puisse modifier la stabilité/traduction de l’ARN qui les porte et ainsi promouvoir le développement de maladies. En prenant comme exemple le proto-oncogène c-Myc et la cytokine pro-inflammatoire TNFα, nous allons examiner comment l’altération de la région 3’ UTR du gène contribue à l’apparition de cancers et de pathologies inflammatoires.
Stabilité des ARNm et cancers
La mutation de la séquence 3’UTR d’un gène incluant un ARE peut empêcher la fixation de différents facteurs agissant en trans, que ce soit des petits ARN, des nucléases ou des AUBP. De telles altérations génétiques, conduisant à la dérégulation du niveau d’expression de l’ARNm muté, ont été observées dans plusieurs cancers (revue dans [1, 8, 21, 22]). Le cas du proto-oncogène c-Myc mérite d’être détaillé car il illustre la complexité des mécanismes de dégradation. Plusieurs translocations entraînant la perte de son ARE sont décrites dans des hémopathies malignes humaines (revue dans [21]). De manière surprenante, cependant, la délétion de la région 3’UTR de c-myc (c-mycΔ3’UTR), créée expérimentalement par recombinaison homologue, n’augmente pas la stabilité de l’ARN c-mycΔ3’UTR et aucune tumeur n’est observée chez la souris exprimant l’ARN muté [23]. Ces résultats, en apparence contradictoires, peuvent s’expliquer par la découverte que plusieurs séquences, dont l’activité dépend du contexte cellulaire, contrôlent la stabilité de l’ARN c-myc. En effet, cet ARN contient dans sa région codante (CRD) une séquence reconnue par la protéine CRD-BP qui la protège, si elle est exprimée, de l’attaque d’endonucléases (revue dans [24]). Comme CRD-BP n’est pas synthétisée dans les tissus sains de la souris adulte, l’ARN c-myc subit l’attaque d’endonucléases, même s’il est délété de son ARE, comme c’est le cas de l’allèle c-mycΔ3’UTR. En revanche, dans les cellules cancéreuses, CRD-BP est fortement exprimée, en particulier dans les cellules dans lesquelles la voie β-caténine/Tcf est activée [25]. Ainsi, dans les hémopathies malignes dans lesquelles la 3’ UTR de c-myc est délétée, l’expression de CRD-BP contribuerait à l’accumulation de l’ARN c-myc déjà stabilisé par la perte de son ARE (Figure 1). Cet exemple illustre bien comment plusieurs régions d’un même ARN pourraient agir, en fonction du contexte cellulaire, indépendamment ou en synergie, pour garantir la destruction rapide d’un ARN lorsqu’elle est nécessaire. Pour l’ARN c-myc dans les cellules cancéreuses, la délétion de la 3’ UTR et l’accumulation de CRD-BP déjouent irrémédiablement ces astucieux plans de surveillance.
Surexpression d’AUBP et cancers
Si la délétion des gènes codant pour des AUBP n’a pas été décrite dans des pathologies humaines, la surexpression d’AUBP est mentionnée dans des stades avancés de cancers. De manière frappante, HuR est l’AUBP la plus souvent citée (revue dans [21]). Son implication dans une grande variété de tumeurs repose probablement sur un ensemble de caractéristiques qui en font une AUBP unique : (1) c’est une protéine dont l’expression est ubiquitaire ; (2) c’est la seule AUBP « stabilisante » parmi les AUBP décrites ; (3) sa fixation à l’ARE est moins stricte que celle d’autres AUBP, AUF1 par exemple, qui requiert en dehors de l’ARE un contexte nucléotidique particulier [26, 27], et ses ARN cibles sont donc très nombreux. L’implication directe d’HuR dans le développement de cancers est illustrée par les travaux du groupe de Gorospe sur un modèle cellulaire de carcinome colorectal, les cellules RKO. L’injection sous-cutanée de ces cellules à des souris nude provoque l’apparition rapide de tumeurs. Mais, si l’on diminue l’expression d’HuR par l’utilisation de petits ARN interférents, le développement tumoral est beaucoup plus lent et s’accompagne d’une réduction de la stabilité des ARNm cycline A et cycline B1, deux cibles d’HuR jouant une rôle clef dans la prolifération cellulaire [21].
AUBP, ARE et maladies inflammatoires
La plupart des cytokines et chimiokines produites au cours de la réponse immune sont codées par des ARN très instables qui contiennent dans leur région 3’ UTR un ARE caractérisé par la réitération de l’heptamère A/UAUUUAA/U (Figure 1). C’est le cas par exemple des cytokines pro-inflammatoires TNFα, IL-1β, IL-6, IL-8 et GM-CSF, des médiateurs de l’inflammation tels que Cox2, et des cytokines anti-inflammatoires IL-10, dont la synthèse est requise pour contrecarrer l’effet pathogène à long terme des cytokines pro-inflammatoires. Cela explique pourquoi l’ARE et les AUBP jouent un rôle fondamental dans la régulation de l’expression des gènes de la réponse inflammatoire [28]. Très peu de données sont disponibles sur des mutations/délétions naturelles de leurs ARE à l’origine de pathologies inflammatoires. Cependant, la délétion expérimentale chez la souris de l’ARE du TNFα atteste de son importance in vivo. En effet, les macrophages des souris TNFα ΔARE, dans lesquelles l’ARE a été délétée par mutagenèse dirigée, expriment un ARNm TNFα constitutivement stable entraînant une production spontanée de TNFα. Les souris porteuses de cette mutation développent une arthrite chronique inflammatoire et une inflammation intestinale (IBD) qui ressemble à la maladie de Crohn chez l’homme [29]. De manière remarquable, les souris mutantes (KO) qui n’expriment pas l’AUBP TTP développent les mêmes symptômes - cachexie, arthrite et dermatite accompagnées de la synthèse d’auto-anticorps - que les souris TNFαΔARE [30]. Cela s’explique très bien par le fait que la cible principale de TTP, l’ARN TNFα, n’est plus dégradée, entraînant une expression constitutive de la protéine TNFα par les macrophages. Il est probable que la stabilisation d’ARNm codant d’autres cytokines, telles que le GM-CSF, dont l’ARE fixe également TTP [31], contribue à l’hyperplasie myéloïde caractéristique des souris TTP−/− [32]. Cette hypothèse est confortée par l’observation d’une prolifération anormale et létale de macrophages et de granulocytes dans des embryons transgéniques exprimant un allèle GM-CSFΔARE dépourvu de l’ARE et ne fixant donc plus TTP [33].
La comparaison des phénotypes observés dans les souris KO pour TTP et dans les souris dans lesquelles d’autres AUBP ont été expérimentalement inactivées par recombinaison homologue est très enrichissante car elle révèle la spécificité des interactions cis/trans. En effet, alors que l’ARN TNFα peut fixer in vitro de nombreuses AUBP, telles que HuR, TTP, AUF1, KSRP, TIA-1 et TIAR (Figure 1), seule l’inactivation de TTP provoque une réaction inflammatoire constitutive. L’inactivation d’AUF1 est normalement sans effet mais lorsque la réaction inflammatoire est expérimentalement stimulée (par exemple par traitement des souris avec du LPS qui mime une infection bactérienne à Gram-), les souris KO AUF1−/− font un choc endotoxique létal accompagné d’une surproduction de TNFα et d’IL-1β due à la stabilisation de leurs ARN à ARE [34]. De la même manière, les souris TIA-1- /- ne développent pas spontanément de maladie. Mais, lorsque leurs macrophages péritonéaux sont traités in vitro par le LPS, la synthèse de TNFα et de Cox2 est accrue. Cependant, dans ce cas, il n’y a pas de modification du niveau d’expression des ARNm correspondants, suggérant que TIA-1 n’agit pas au niveau de la stabilité des ARNm, comme le fait TTP, mais de leur traduction [35]. L’action conjointe de TTP et de TIA-1 sur le niveau d’expression de cytokines inflammatoires est démontrée in vivo par l’analyse comparative des souris simples ou doubles KO pour ces deux gènes : les souris TTP−/− développent une arthrite plus sévère que les souris TIA-1−/− mais moins sévère que celle que l’on observe dans les souris doubles mutantes TTP−/− et TIA−/− [36]. Un autre exemple, tiré cette fois-ci de l’analyse de souris transgéniques surexprimant la protéine HuR, confirme le rôle et la complexité de la régulation des cytokines par les AUBP. Comme on s’y attend compte tenu de la fonction stabilisante de cette AUBP, il y a bien une augmentation de la stabilité des ARN TNFα et Cox2 dans les cellules myéloïdes qui surexpriment le transgène. Cependant, la réponse inflammatoire n’est pas activée car cette surexpression, en synergie avec la protéine TIA-1, inhibe la traduction de ces ARNm [37].
Des modifications post-traductionnelles d’AUBP aux maladies
On connait depuis longtemps l’importance de l’activation de voies de signalisation dans la régulation post-transcriptionnelle de la biosynthèse des cytokines en réponse au stress [38]. L’implication précise de la voie MAPK p38 dans l’induction de l’expression du TNFα a été mise en évidence par l’utilisation de souris dans lesquelles une des kinases activées par la kinase p38 est inactivée, les souris MK2−/− (Figure 3). Le traitement des souris MK2−/− par le LPS ne provoque pas de réponse inflammatoire car il n’y a pas de synthèse de TNFα. TTP est faiblement exprimé dans leurs macrophages et, contrairement à ce qu’on observe dans les souris sauvages, il n’est pas phosphorylé. Le modèle proposé est que l’activation de la voie p38, via la kinase MK2, provoque la phosphorylation de TTP, favorisant ainsi à la fois sa stabilisation, sa liaison à la protéine 14-3-3 et la réduction de son affinité pour l’ARE. Il en résulte une accumulation transitoire, tant que perdure l’activation de la voie de signalisation, des ARN TNFα [39]. Dans les souris MK2−/−, la faible expression de TTP entraîne bien une accumulation des transcrits TNFα, mais ils ne sont pas traduits. Ces résultats suggèrent que la voie p38 a deux effets sur l’expression de TNFα qui reposent tous les deux sur la présence de l’ARE : un rôle sur l’accumulation des transcrits via la modulation de l’interaction ARE/TTP, et un rôle sur leur traduction via un (ou des) facteur(s) qu’il reste à caractériser. Un de ces facteurs pourrait être l’inhibiteur traductionnel TIA-1 puisque son ablation génétique provoque la traduction de l’ARN TNFα sans affecter son abondance [35] (Figure 3).
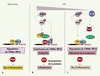 |
Figure 3. Modèle de la cinétique d’action de l’ARE du TNFaen réponse à l’activation de la voie MAP-kinase p38. A. Dans des conditions normales, l’AUBP déstabilisante TTP se fixe sur l’ARE du TNFα ce qui provoque sa dégradation. B. L’activation de la voie p38 entraîne une phosphorylation de TTP qui « libère » l’ARE du TNFα, le rendant éventuellement accessible à des protéines stabilisantes comme HuR. Les transcrits accumulés sont activement traduits, ce qui présuppose la levée de l’inhibition traductionnelle exercée par TIA-1/TIAR. L’activation de p38 pourrait favoriser la phosphorylation de TIA-1/TIAR. Bien que sa phosphorylation n’altère pas sa liaison à l’ARE [35], elle pourrait modifier sa capacité à inhiber la traduction. Alternativement TIA-1/TIAR pourrait être en quantité trop faible pour réprimer la traduction de l’ensemble des molécules d’ARNm TNFα synthétisées lors de la stimulation des cellules par le LPS (lipopolysaccharide). Lorsque l’activation de la voie s’estompe, le stock de TTP -préservé de la dégradation par sa liaison à la protéine 14-3-3- est déphosphorylé et peut à nouveau fixer l’ARE entraînant une dégradation de l’ARN. La quantité de l’ARN est alors diminuée rétablissant ainsi l’équilibre stœchiométrique entre TIA-1/TIAR et sa cible ARN. C. Parallèlement à l’activation de la voie p38, IL-10, un inhibiteur clé de la réponse inflammatoire, est synthétisé. Elle exerce un effet négatif sur la traduction du TNFα en diminuant la synthèse d’HuR[42]. |
L’implication des voies de signalisation dans la régulation de l’activité des AUBP ne se limite certainement pas au contrôle de l’homéostasie du système immunitaire. En effet, la découverte récente qu’AUF1 a pour partenaire une kinase oncogénique suggère que des modifications post-traductionnelles d’AUBP puissent également être impliquées dans l’apparition de cancers [40]. Les auteurs montrent dans cette étude qu’AUF1 s’associe à la tyrosine kinase NMP-ALK caractéristique des lymphomes anaplasiques à grandes cellules et que son hyperphosphorylation s’accompagne d’une augmentation de la stabilisation d’ARNm à ARE, tels que ceux codant pour c-Myc et les cyclines D1, A2 et B1. AUF1 se trouve associé avec la tyrosine kinase oncogénique dans des granules cytoplasmiques. Ces granules pourraient, à l’instar des P bodies et des granules de stress, jouer un rôle dans le contrôle de la dégradation et/ou du stockage des ARNm et ainsi contribuer au phénotype tumoral des cellules exprimant la kinase oncogénique. Ainsi non seulement le niveau d’expression des AUBP, mais également leur localisation sub-cellulaire et leurs modifications post-traductionnelles, déterminent leur capacité à interagir avec l’ARE et à moduler la stabilité de l’ARN qui le porte.

Conclusion
On ne fait qu’entrevoir l’importance des modifications post-traductionnelles dans la régulation de l’expression des ARN à ARE au cours de processus physiologiques. Leur étude dans des processus pathologiques nécessite l’élaboration d’outils performants, comme des inhibiteurs des différentes étapes des voies de signalisation et des anticorps capables de reconnaitre spécifiquement une modification post-traductionnelle précise, qui font cruellement défaut actuellement. La découverte que la stabilité et la traduction de nombreux ARNm sont fréquemment dérégulées dans des cancers et maladies inflammatoires ouvre de nombreuses perspectives tant du point de vue fondamental que thérapeutique.
Références
- Ross J. mRNA stability in mammalian cells. Microbiol Rev 1995; 59 : 423–450. [Google Scholar]
- Gillet R, Felden B. Lost in translation : le déblocage des ribosomes bactériens par le mécanisme de trans-traduction. Med Sci (Paris) 2007; 23 : 633–9 [Google Scholar]
- Shaw G, Kamen R. A conserved AU sequence from the 3’ untranslated region of GM-CSF mRNA mediates selective mRNA degradation. Cell 1986; 46 : 659–67. [Google Scholar]
- Bakheet T, Williams BR, Khabar KS. ARED 3.0: the large and diverse AU-rich transcriptome. Nucleic Acids Res 2006; 34 : D111–4. [Google Scholar]
- Chen CYA, Shyu AB. AU-rich elements:characterization and importance in mRNA degradation. Trends Biochem Sci 1995; 20 : 465–70. [Google Scholar]
- Puig S, Askeland E, Thiele DJ. Coordinated remodeling of cellular metabolism during iron deficiency through targeted mRNA degradation. Cell 2005; 120 : 99–110. [Google Scholar]
- Guhaniyogi J, Brewer G. Regulation of mRNA stability in mammalian cells. Gene 2001; 265 : 11–23. [Google Scholar]
- Barreau C, Paillard L, Osborne HB. AU-rich elements and associated factors: are there unifying principles ? Nucleic Acids Res 2005; 33 : 7138–50. [Google Scholar]
- Bevilacqua A, Ceriani MC, Capaccioli S, Nicolin A. Post-transcriptional regulation of gene expression by degradation of messenger RNAs. J Cell Physiol 2003; 195 : 356–72. [Google Scholar]
- Gamberi C, Johnstone O, Lasko P. Drosophila RNA binding proteins. Int Rev Cytol 2006; 248 : 43–139. [Google Scholar]
- Liao B, Hu Y, Brewer G. Competitive binding of AUF1 and TIAR to MYC mRNA controls its translation. Nat Struct Mol Biol 2007; 14 : 511–8. [Google Scholar]
- Lal A, Mazan-Mamczarz K, Kawai T, et al. Concurrent versus individual binding of HuR and AUF1 to common labile target mRNAs. EMBO J 2004; 23 : 3092–102. [Google Scholar]
- Camier S, Séraphin B. Détruisez ce message (ARN) après l’avoir lu ! Med Sci (Paris) 2007; 23 : 850–6. [Google Scholar]
- Chen CY, Gherzi R, Ong SE, et al. AU binding proteins recruit the exosome to degrade ARE-containing mRNAs. Cell 2001; 107 : 451–64. [Google Scholar]
- Lin WJ, Duffy A, Chen CY. Localization of AU-rich element-containing mRNA in cytoplasmic granules containing exosome subunits. J Biol Chem 2007; 282 : 19958–68. [Google Scholar]
- Parker R, Sheth U. P bodies and the control of mRNA translation and degradation. Mol Cell 2007; 25 : 635–46. [Google Scholar]
- Eulalio A, Behm-Ansmant I, Izaurralde E. P bodies: at the crossroads of post-transcriptional pathways. Nat Rev Mol Cell Biol 2007; 8 : 9–22. [Google Scholar]
- Kedersha N, Stoecklin G, Ayodele M, et al. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J Cell Biol 2005; 169 : 871–84. [Google Scholar]
- Bhattacharyya SN, Habermacher R, Martine U, et al. Relief of microRNA-Mediated Translational Repression in Human Cells Subjected to Stress. Cell 2006; 125 : 1111–24. [Google Scholar]
- Jing Q, Huang S, Guth S, et al. Involvement of microRNA in AU-rich element-mediated mRNA instability. Cell 2005; 120 : 623–34. [Google Scholar]
- Lopez de Silanes L, Quesada P, Esteller M. Aberrant regulation of messenger RNA 3’-untranslated region in human cancer. Cell Oncol 2007; 29 : 1–17. [Google Scholar]
- Wiemer EA. The role of microRNAs in cancer: no small matter. Eur J Cancer 2007; 43 : 1529–44. [Google Scholar]
- Langa F, Lafon I, Vandormael-Pournin S, et al. Healthy mice with an altered c-myc gene: role of the 3’ untranslated region revisited. Oncogene 2001; 20 : 4344–53. [Google Scholar]
- Sparanese D, Lee CH. CRD-BP shields c-myc and MDR-1 RNA from endonucleolytic attack by a mammalian endoribonuclease. Nucleic Acids Res 2007; 35 : 1209–21. [Google Scholar]
- Noubissi FK, Elcheva I, Bhatia N, et al. CRD-BP mediates stabilization of betaTrCP1 and c-myc mRNA in response to beta-catenin signalling. Nature 2006; 441 : 898–901. [Google Scholar]
- Blaxall BC, Pende A, Wu SC, Port JD. Correlation between intrinsic mRNA stability and the affinity of AUF1 (hnRNP D) and HuR for A + U-rich mRNAs. Mol Cell Biochem 2002; 232 : 1–11. [Google Scholar]
- Fialcowitz EJ, Brewer BY, Keenan BP, Wilson GM. A hairpin-like structure within an AU-rich mRNA-destabilizing element regulates trans-factor binding selectivity and mRNA decay kinetics. J Biol Chem 2005; 280 : 22406–17. [Google Scholar]
- Espel E. The role of the AU-rich elements of mRNAs in controlling translation. Semin Cell Dev Biol 2005; 16 : 59–67. [Google Scholar]
- Kontoyiannis D, Pasparakis M, Pizarro TT, et al. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity 1999; 10 : 387–98. [Google Scholar]
- Taylor GA, Carballo E, Lee DM, et al. A pathogenetic role for TNF alpha in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity 1996; 4 : 445–54. [Google Scholar]
- Carballo E, Lai W, Blackshear P. Evidence that tristetraprolin is a physiological regulator of granulocyte-macrophage colony-stimulating factor messenger RNA deadenylation and stability. Blood 2000; 95 : 1891–9. [Google Scholar]
- Ogilvie RL, Abelson M, Hau HH, et al. Tristetraprolin down-regulates IL-2 gene expression through AU-rich element-mediated mRNA decay. J Immunol 2005; 174 : 953–61. [Google Scholar]
- Houzet L, Morello D, Defrance P, et al. Regulated control by GM-CSF AU-rich element during mouse embryogenesis. Blood 2001; 9 : 1281–8. [Google Scholar]
- Lu JY, Sadri N, Schneider RJ. Endotoxic shock in AUF1 knockout mice mediated by failure to degrade proinflammatory cytokine mRNAs. Genes Dev 2006; 20 : 3174–84. [Google Scholar]
- Piecyk M, Wax S, Beck AR, et al. TIA-1 is a translational silencer that selectively regulates the expression of TNF-alpha. EMBO J 2000; 19 : 4154–63. [Google Scholar]
- Phillips K, Kedersha N, Shen L, et al. Arthritis suppressor genes TIA-1 and TTP dampen the expression of tumor necrosis factor alpha, cyclooxygenase 2, and inflammatory arthritis. Proc Natl Acad Sci USA 2004; 101 : 2011–6. [Google Scholar]
- Katsanou V, Papadaki O, Milatos S, et al. HuR as a negative posttranscriptional modulator in inflammation. Mol Cell 2005; 19 : 777–89. [Google Scholar]
- Saklatvala J. The p38 MAP kinase pathway as a therapeutic target in inflammatory disease. Curr Opin Pharmacol 2004; 4 : 372–7. [Google Scholar]
- Hitti E, Iakovleva T, Brook M, et al. Mitogen-activated protein kinase-activated protein kinase 2 regulates tumor necrosis factor mRNA stability and translation mainly by altering tristetraprolin expression, stability, and binding to adenine/uridine-rich element. Mol Cell Biol 2006; 26 : 2399–407. [Google Scholar]
- Fawal M, Armstrong F, Ollier S, et al. « Liaison dangereuse » between AUF1/hnRNPD and the oncogenic tyrosine kinase NPM-ALK. Blood 2006; 108 : 2780–8. [Google Scholar]
- Tafech A, Bennett WR, Mills F, Lee CH. Identification of c-myc coding region determinant RNA sequences and structures cleaved by an RNase1-like endoribonuclease. Biochim Biophys Acta 2007; 1769 : 49–60. [Google Scholar]
- Rajasingh J, Bord E, Luedemann C, et al. IL-10-induced TNF-alpha mRNA destabilization is mediated via IL-10 suppression of p38 MAP kinase activation and inhibition of HuR expression. Faseb J 2006; 20 : 2112–4. [Google Scholar]
Liste des figures
|
Figure 1. Diversité des séquences ARE et des AUBP avec lesquelles elles interagissent. La classification initiale des ARE en trois catégories [5] s’est enrichie de subdivisions de la catégorie II en 4 groupes suivant le nombre de pentamères AUUUA [4]. L’ARE de l’ARN c-myc (nt 2061-2141 NM_002467) illustre la catégorie I et celle du TNFa la catégorie II, groupe 3 (nt n° 1311-1385 du gène TNFα NM_000594). À titre de comparaison, l’ARE de c-jun, un exemple de la catégorie III qui ne contient pas de pentamère mais une région riche en uridines ainsi que plusieurs motifs GUUUG est indiqué (nt 2401-2542 du gène c-jun J04115). Deux AUUUA dispersés dans un contexte riche en uridines sont présents dans l’ARE de c-myc, alors que le pentamère se répète 8 fois, dont 6 de manière chevauchante, pour l’ARE du TNFa. Les AUBP interagissant avec ces 2 dernières séquences sont indiquées (en jaune les AUBP déstabilisantes, en rouge foncé la seule AUBP stabilisante et en bleu les régulateurs traductionnels). miR16 contient la séquence UAAAUAUU et peut donc s’apparier à plusieurs sites de l’ARE du TNFα [20]. La région codée par l’exon 3 de c-myc contient la séquence CRD reconnue par la protéine CRD-BP qui la protège d’action d’endonucléases (pour revue, voir [41]) et dont l’expression est activée par la voie β-caténine/Tcf. |
| Dans le texte | |
|
Figure 2. Localisation des ARNm dans divers granules cytoplasmiques. Les ARNm sont localisés dans différentes structures cytoplasmiques où ils sont stockés (granules de stress et P bodies) ou dégradés (exosomes bodies et P bodies). En fonction du stimulus cellulaire, les ARNm associés à différentes protéines peuvent être libérés de ces granules et activement traduits dans les polysomes (d’après [16–19]). |
| Dans le texte | |
|
Figure 3. Modèle de la cinétique d’action de l’ARE du TNFaen réponse à l’activation de la voie MAP-kinase p38. A. Dans des conditions normales, l’AUBP déstabilisante TTP se fixe sur l’ARE du TNFα ce qui provoque sa dégradation. B. L’activation de la voie p38 entraîne une phosphorylation de TTP qui « libère » l’ARE du TNFα, le rendant éventuellement accessible à des protéines stabilisantes comme HuR. Les transcrits accumulés sont activement traduits, ce qui présuppose la levée de l’inhibition traductionnelle exercée par TIA-1/TIAR. L’activation de p38 pourrait favoriser la phosphorylation de TIA-1/TIAR. Bien que sa phosphorylation n’altère pas sa liaison à l’ARE [35], elle pourrait modifier sa capacité à inhiber la traduction. Alternativement TIA-1/TIAR pourrait être en quantité trop faible pour réprimer la traduction de l’ensemble des molécules d’ARNm TNFα synthétisées lors de la stimulation des cellules par le LPS (lipopolysaccharide). Lorsque l’activation de la voie s’estompe, le stock de TTP -préservé de la dégradation par sa liaison à la protéine 14-3-3- est déphosphorylé et peut à nouveau fixer l’ARE entraînant une dégradation de l’ARN. La quantité de l’ARN est alors diminuée rétablissant ainsi l’équilibre stœchiométrique entre TIA-1/TIAR et sa cible ARN. C. Parallèlement à l’activation de la voie p38, IL-10, un inhibiteur clé de la réponse inflammatoire, est synthétisé. Elle exerce un effet négatif sur la traduction du TNFα en diminuant la synthèse d’HuR[42]. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.