")
")
| Issue |
Med Sci (Paris)
Volume 24, Number 3, Mars 2008
|
|
|---|---|---|
| Page(s) | 226 - 228 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/2008243226 | |
| Published online | 15 mars 2008 | |
Un transporteur d’anions est essentiel à la mobilité des spermatozoïdes
Testis anion transporteur 1 and male fertility
1
Institut Cochin, Inserm U567-UMR 8104 CNRS, 24, rue du Faubourg Saint-Jacques, 75014 Paris, France
2
Université Paris XI, Laboratoire d’Andrologie, CHU de Bicêtre, 78, avenue du Général Leclerc, 94275 Le Kremlin Bicêtre-Cedex, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
La famille SLC26 de transporteurs d’anions
La famille Slc26 est une famille de transporteurs d’anions monovalents et divalents conservée au cours de l’évolution. Chez l’homme, quatre de ces transporteurs, SLC26A2, A3, A4 et A5, sont la cible de mutations « perte de fonction » responsables d’affections diverses : respectivement chondrodysplasie, diarrhée chlorurée congénitale, syndrome de Pendred et surdité. Bien que cliniquement distinctes, ces maladies résultent toutes d’un défaut de transport d’anions dans les tissus où les transporteurs SLC26 sont exprimés, le plus souvent de manière spécifique [1, 2].
Ainsi, les mutations du gène SLC26A2 sont responsables de diverses dysplasies osseuses et dans les cas les plus extrêmes, elles conduisent à un nanisme accompagné de sévères atteintes squelettiques (pied-bot, scoliose…). Chez les patients porteurs de ces mutations, l’activité du transporteur de sulfate SLC26A2/DTDST, préférentiellement exprimé au niveau du cartilage, est abolie. On observe une réduction du transport de sulfate dans les chondrocytes conduisant à une hyposulfatation des protéoglycanes de la matrice extracellulaire du cartilage [3–5]. De manière analogue, les mutations du gène SLC26A4 dont l’expression est restreinte à l’oreille interne (cochlée) et à la thyroïde, sont à l’origine du syndrome de Pendred ; dans ce cas, les patients présentent une surdité bilatérale liée à une malformation de la cochlée et un goitre thyroïdien consécutif à un défaut d’organification de l’iodure au niveau thyroïdien [6,7].
Notre équipe a précédemment isolé la protéine TAT1 (testis anion transporteur 1) comme un nouveau membre de la famille SLC26 [8]. TAT1/SLC26A8 s’exprime de façon exclusive au niveau du testicule et spécifiquement dans la lignée germinale mâle, dès le stade spermatocyte. La protéine TAT1 est présente à la surface des spermatocytes et des spermatides ; dans les spermatozoïdes, elle est localisée spécifiquement dans la région de l’annulus, une structure fibrillaire sous-membranaire située à la jonction des pièces intermédiaire et principale du flagelle (Figure 1). In vitro, la protéine TAT1 transporte les anions sulfate, chlorure et oxalate [8,9].
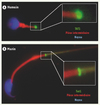 |
Figure 1. Localisation sub-cellulaire de la protéine TAT1 sur les spermatozoïdes humains (A) et murins (B). La protéine TAT1 (marquage vert) est localisée au niveau de l’annulus, structure à la jonction des pièces intermédiaire (marquage rouge) et principale du flagelle. |
Afin d’élucider la fonction physiologique du transporteur TAT1 dans la lignée germinale mâle, notre équipe a créé puis caractérisé un modèle d’inactivation du gène Tat1 par recombinaison homologue chez la souris.
Phénotype induit par l’invalidation du gène Tat1 chez la souris
Dans les tubes séminifères du testicule sont produits des spermatozoïdes structuralement différenciés mais encore incapables de féconder l’ovocyte. Pour acquérir leur potentiel fertilisant, les spermatozoïdes subissent des événements de maturation dans l’épididyme et dans les voies génitales femelles [10]. Le transit épididymaire permet aux spermatozoïdes d’acquérir leur mobilité ; puis dans les voies génitales femelles, il se produit un ensemble de modifications appelé capacitation, qui conduit à l’hyperactivation flagellaire (augmentation de la fréquence et de l’amplitude du battement flagellaire) et à l’acquisition du potentiel de réaction acrosomale nécessaire à l’interaction avec l’ovocyte.
L’inactivation du gène Tat1 par recombinaison homologue chez la souris induit, comme seul phénotype observable, une infertilité des souris mâles Tat1−/−. L’analyse histologique du testicule et de l’épididyme ainsi que le comptage des réserves spermatiques indiquent que la spermatogenèse des mâles Tat1−/− se déroule normalement et permet la production d’une quantité normale de spermatozoïdes. En revanche, les mesures de mobilité effectuées par le système CASA (computer aided sperm analysis) révèlent une absence quasi complète de mobilité des spermatozoïdes Tat1−/− (asthénozoospermie) [11].
Par ailleurs, le flagelle des spermatozoïdes Tat1−/− présente un amincissement à la jonction des pièces intermédiaire et principale qui conduit à un repliement du flagelle lors du transit épididymaire (Figure 2A) et entraîne la rupture des structures axonémales observable en microscopie électronique [11].
 |
Figure 2. Défaut de structure et absence d’hyperactivation des spermatozoïdes Tat1−/−. A. Contrairement aux spermatozoïdes Tat1+/+ (à gauche), les spermatozoïdes Tat1−/− présentent une disjonction entre la pièce intermédiaire et la pièce principale (au centre) conduisant à un repliement du flagelle à 180° (à droite). B. L’incubation en milieu capacitant des spermatozoïdes Tat1−/− ne permet pas d’induire l’hyperphosphorylation protéique accompagnant le processus d’hyperactivation flagellaire (détection par Western blot avec un anticorps dirigé contre les phospho-tyrosines). |
Il est possible d’évaluer, in vitro, le potentiel fertilisant des spermatozoïdes en analysant le profil d’hyperphosphorylation accompagnant le processus d’hyperactivation flagellaire et la capacité des spermatozoïdes à réaliser la réaction acrosomale dans un milieu capacitant artificiel. Nos études indiquent que les spermatozoïdes Tat1−/− ne sont pas capables de réaliser l’hyperphosphorylation des protéines flagellaires (Figure 2B) ; en revanche, la majorité d’entre eux (70 %) est capable d’effectuer la réaction acrosomique [11], ce qui suggère une altération des événements de maturation au niveau flagellaire.
Perspectives dans les domaines de la contraception et des infertilités masculines
L’ensemble de nos résultats indique clairement que la protéine Tat1/Slc26a8 est essentielle à la fertilité des souris mâles et qu’elle contrôle les processus de mobilité et d’hyperactivation flagellaire. Ces processus font intervenir plusieurs facteurs biochimiques et ioniques ; ainsi le rôle déterminant des influx d’ions bicarbonate et calcium a été établi depuis plusieurs années [12]. Il est possible que le transporteur Tat1 intervienne dans la régulation des échanges ioniques accompagnant les processus de mobilité et d’hyperactivation. Si tel est le cas, la protéine Tat1 pourrait constituer une cible pharmacologique intéressante dans le cadre du développement de contraceptifs masculins. En effet, du fait de son expression exclusive dans les cellules germinales mâles (d’après l’analyse de 17 tissus adultes humains [8, 9]) et de sa localisation à la membrane plasmique de ces cellules, la protéine TAT1 constitue une cible accessible à d’éventuels inhibiteurs pharmacologiques.
Par ailleurs, plusieurs équipes ont récemment créé des modèles d’invalidation des gènes Slc26 chez la souris dans le but d’éclaircir les fonctions physiologiques de ces transporteurs. Ces modèles se sont révélés particulièrement intéressants dans la mesure où ils reproduisent la plupart des symptômes observés dans les maladies humaines correspondantes [13–16]. Les souris mâles Tat1−/− constituent de ce fait un modèle pertinent pour l’investigation du rôle de la protéine Tat1 dans le contrôle de la fertilité masculine. Au vu de ces résultats, il est en particulier légitime de rechercher l’implication de mutations du gène TAT1 dans les asthénozoospermies humaines. Les causes génétiques des infertilités masculines restent encore très peu connues, et ce travail pourrait donc contribuer à une meilleure connaissance étiologique de cette pathologie.
Références
- Everett LA, Green ED. A family of mammalian anion transporters and their involvement in human genetic diseases. Hum Mol Genet 1999; 8 : 1883–91. [Google Scholar]
- Dawson PA, Markovich D. Pathogenetics of the human SLC26 transporters. Curr Med Chem 2005; 12 : 385–96. [Google Scholar]
- Hastbacka J, De la Chapelle A, Mahtani MM, et al. The diastrophic dysplasia gene encodes a novel sulfate transporter: positional cloning by fine-structure linkage disequilibrium mapping. Cell 1994; 78 : 1073–87. [Google Scholar]
- Karniski LP. Mutations in the diastrophic dysplasia sulfate transporter (DTDST) gene: correlation between sulfate transport activity and chondrodysplasia phenotype. Hum Mol Genet 2001; 10 : 1485–90. [Google Scholar]
- Rossi A, Bonaventure J, Delezoide AL, et al. Undersulfation of proteoglycans synthesized by chondrocytes from a patient with achondrogenesis type 1B homozygous for an L483P substitution in the diastrophic dysplasia sulfate transporter. J Biol Chem 1996; 271 : 18456–64. [Google Scholar]
- Sheffield VC, Kraiem Z, Beck JC, et al. Pendred syndrome maps to chromosome 7q21-34 and is caused by an intrinsic defect in thyroid iodine organification. Nat Genet 1996; 12 : 424–6. [Google Scholar]
- Coyle B, Coffey R, Armour JA, et al. Pendred syndrome (goitre and sensorineural hearing loss) maps to chromosome 7 in the region containing the nonsyndromic deafness gene DFNB4. Nat Genet 1996; 12 : 421–3. [Google Scholar]
- Toure A, Morin L, Pineau C, et al. Tat1, a novel sulfate transporter specifically expressed in human male germ cells and potentially linked to rhogtpase signaling. J Biol Chem 2001; 276 : 20309–15. [Google Scholar]
- Lohi H, Kujala M, Makela S, et al. Functional characterization of three novel tissue-specific anion exchangers SLC26A7, -A8, and -A9. J Biol Chem 2002; 277 : 14246–54. [Google Scholar]
- Yanagimachi R. Fertility of mammalian spermatozoa: its development and relativity. Zygote 1994; 2 : 371–2. [Google Scholar]
- Toure A, Lhuillier P, Gossen JA, et al. The Testis Anion Transporter 1 (Slc26a8) is required for sperm terminal differentiation and male fertility in the mouse. Hum Mol Genet 2007; 16 : 1783–93. [Google Scholar]
- Visconti PE, Westbrook VA, Chertihin O, et al. Novel signaling pathways involved in sperm acquisition of fertilizing capacity. J Reprod Immunol 2002; 53 : 133–50. [Google Scholar]
- Forlino A, Piazza R, Tiveron C, et al. A diastrophic dysplasia sulfate transporter (SLC26A2) mutant mouse: morphological and biochemical characterization of the resulting chondrodysplasia phenotype. Hum Mol Genet 2005; 14 : 859–71. [Google Scholar]
- Everett LA, Belyantseva IA, Noben-Trauth K, et al. Targeted disruption of mouse Pds provides insight about the inner-ear defects encountered in Pendred syndrome. Hum Mol Genet 2001; 10 : 153–61. [Google Scholar]
- Wang Z, Wang T, Petrovic S, et al. Renal and intestinal transport defects in Slc26a6-null mice. Am J Physiol Cell Physiol 2005; 288 : C957–65. [Google Scholar]
- Cheatham MA, Huynh KH, Gao J, et al. Cochlear function in Prestin knockout mice. J Physiol 2004; 560 : 821–30. [Google Scholar]
© 2008 médecine/sciences - Inserm / SRMS
Liste des figures
|
Figure 1. Localisation sub-cellulaire de la protéine TAT1 sur les spermatozoïdes humains (A) et murins (B). La protéine TAT1 (marquage vert) est localisée au niveau de l’annulus, structure à la jonction des pièces intermédiaire (marquage rouge) et principale du flagelle. |
| Dans le texte | |
|
Figure 2. Défaut de structure et absence d’hyperactivation des spermatozoïdes Tat1−/−. A. Contrairement aux spermatozoïdes Tat1+/+ (à gauche), les spermatozoïdes Tat1−/− présentent une disjonction entre la pièce intermédiaire et la pièce principale (au centre) conduisant à un repliement du flagelle à 180° (à droite). B. L’incubation en milieu capacitant des spermatozoïdes Tat1−/− ne permet pas d’induire l’hyperphosphorylation protéique accompagnant le processus d’hyperactivation flagellaire (détection par Western blot avec un anticorps dirigé contre les phospho-tyrosines). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.