")
")
| Issue |
Med Sci (Paris)
Volume 24, Number 1, Janvier 2008
|
|
|---|---|---|
| Page(s) | 25 - 28 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/200824125 | |
| Published online | 15 January 2008 | |
Identification d’une voie synaptique associée à l’autisme
Alterations in synapsis formation and function in autism disorders
1
Laboratoire de Génétique Humaine et Fonctions Cognitives, Institut Pasteur, 25, rue du Docteur Roux, 75724 Paris Cedex 15, France
2
Inserm U513, Neurobiologie et Psychiatrie, Faculté de Médecine, 8, rue du Général Sarrail, 94010 Créteil Cedex, France
3
Inserm U513, Service de Psychiatrie Adulte, Hôpitaux Albert Chenevier et Henri Mondor, Assistance Publique-Hôpitaux de Paris, 40, rue de Mesly, 94000 Créteil, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
L’autisme est caractérisé par un déficit de l’interaction sociale réciproque, un déficit qualitatif de la communication verbale et non verbale, et un répertoire de comportements restreints, répétitifs et stéréotypés. Ces troubles apparaissent chez l’enfant avant l’âge de trois ans et affectent environ un enfant sur 166 pour « les troubles du spectre autistique » (TSA) et un enfant sur 1 000 pour l’autisme typique dit « autisme de Kanner ». Des résultats récents obtenus sur les protéines synaptiques NLGN3, NLGN4, SHANK3 et NRXN1 suggèrent fortement l’implication de la formation et de la maturation des synapses dans l’étiologie de l’autisme.
Défini par Léo Kanner en 1943 comme un « trouble du contact affectif », le syndrome autistique est classé parmi les troubles envahissants du développement. Il est caractérisé par un déficit de l’interaction sociale réciproque, un déficit qualitatif de la communication verbale et non verbale, et un répertoire de comportements restreints, répétitifs et stéréotypés. De manière indépendante, Hans Asperger décrit en 1944 un groupe d’enfants qui ont un développement normal de l’intelligence et du langage, mais qui présentent des comportements proches de l’autisme avec une déficience des interactions sociales et de la communication. Ainsi, l’autisme n’est certainement pas un syndrome homogène, mais plutôt un ensemble de conditions que l’on nomme maintenant « les troubles du spectre autistique » (TSA). Ces troubles apparaissent chez l’enfant avant l’âge de trois ans et affectent environ un enfant sur 166 pour les TSA et un enfant sur 1 000 pour l’autisme typique dit « autisme de Kanner », avec un risque plus élevé pour les garçons (4 : 1). Chez 10-25 % des personnes atteintes, l’autisme est associé à des maladies génétiques connues, comme la sclérose tubéreuse (impliquant les gènes TSC1 et TSC2), le syndrome de l’X fragile (le gène FMR1), et le syndrome de Rett (le gène MeCP2), on parle alors d’autisme syndromique. Cependant, dans la majorité des cas, l’étiologie de l’autisme demeure inconnue. L’agrégation familiale de l’autisme dit de « Kanner » (4,5 % de récurrence dans la fratrie comparé à la prévalence de 0,1 % en population générale) et l’excès important de jumeaux monozygotes concordants (~90 %) par rapport aux jumeaux dizygotes (<5 %) démontrent la forte contribution génétique dans l’autisme non syndromique [1]. À partir des analyses de liaisons et/ou des remaniements chromosomiques observés chez les personnes avec autisme, plusieurs mutations concernant des protéines synaptiques (NLGN3, NLGN4, SHANK3 et NRXN1) ont été identifiées par notre groupe et par le consortium international Autism Genome Project [2].
Des protéines d’adhérence cellulaire à la synapse : les Neuroligines et les Neurexines
En 2003, les premières mutations altérant deux gènes situés sur le chromosome X (NLGN3 et NLGN4) et codant les Neuroligines 3 et 4 ont été mises en évidence chez des personnes avec autisme ou syndrome d’Asperger [3]. Dans deux familles indépendantes, une insertion entraînant l’apparition prématurée d’un codon stop dans NLGN4 et un changement d’acide aminé (R451C) dans NLGN3 ont été identifiés. Les mutations sont transmises par les mères aux deux garçons atteints, dont, pour les deux familles, l’un est atteint d’autisme et l’autre du syndrome d’Asperger. Les analyses fonctionnelles montrent que les mutations altèrent la formation des synapses [4]. En effet, les Neuroligines (NLGN) sont des molécules d’adhésion cellulaire localisées au niveau de la membrane post-synaptique des neurones et sont capables in vitro de déclencher la formation de synapses entre des cellules non neuronales et des neurones en culture. In vivo, l’analyse détaillée de la souris invalidée pour les gènes Nlgn1, Nlgn2 et Nlgn3 montre une diminution sévère de l’activité GABAergique sans modification du nombre de synapses. Ainsi, ces protéines ne sont pas indispensables pour la formation des synapses, mais elles sont néanmoins cruciales pour le fonctionnement des réseaux neuronaux. De plus, une étude récente a montré que les souris portant la mutation R451C dans le gène nlgn3 présentent une augmentation des courants inhibiteurs et un défaut des interactions sociales [5]. Les Neuroligines semblent ainsi déterminantes pour établir un équilibre adéquat entre synapses excitatrices et inhibitrices [6].
Depuis cette première étude dans l’autisme, la recherche de mutations des gènes NLGN3 et NLGN4 a permis d’identifier d’autres mutations, mais ces altérations sont toujours différentes d’une famille à l’autre et ne sont retrouvées que très rarement. À partir des résultats publiés jusqu’à ce jour, il semble que des altérations des gènes NLGN3 et NLGN4 soient présentes chez moins de 1 % des patients (Tableau I). Cependant, ces études ne concernent que les parties codantes des gènes codant les neuroligines et une étude récente rapporte la présence d’un transcrit anormal de NLGN4 dans les lignées lymphoblastoïdes établies à partir d’une fille atteinte d’autisme, sans mutation des sites d’épissage [7]. Ces résultats, s’ils sont répliqués, ouvrent de nouvelles pistes d’études de ces gènes dans l’autisme.
Bilan des études des gènes NLGN3 et NLGN4 dans l’autisme et le retard mental. Asp : syndrome d’Asperger ; RM : retard mental ; TSA : troubles du spectre autistique.
Les neuroligines sont localisées au niveau postsynaptique et interagissent avec les neurexines (NRXN) localisées sur la partie présynaptique des synapses. Très récemment, une étude effectuée par le consortium Autism Genome Project portant sur 1168 familles comportant au moins deux enfants atteints d’autisme, a permis l’identification d’une délétion de novo du gène NRXN1 chez deux sœurs avec autisme [13]. Ces résultats confirment donc l’implication de cette voie synaptique dans l’autisme.
Une protéine d’échafaudage de la synapse : SHANK3
Le syndrome de microdélétion 22q13.3 est caractérisé par une hypotonie néonatale, un retard de développement, un retard sévère voire une absence totale du langage, un retard mental et pour certains cas des traits autistiques [14]. De plus, une translocation 46,XY,t(12 ;22) (q24.1;q13.3) interrompant le gène SHANK3 a été décrite chez un garçon avec un retard de développement, un retard de développement du langage et un retard mental modéré [15]. Parmi les gènes compris dans la délétion, SHANK3 code une protéine synaptique qui interagit avec les Neuroligines et fait le lien entre les récepteurs synaptiques et le cytosquelette d’actine (Figure 1). Une étude approfondie du gène SHANK3 réalisée chez plusieurs enfants atteints d’autisme a permis d’identifier des mutations chez cinq enfants atteints d’autisme ou du syndrome d’Asperger. Dans un premier cas, une délétion d’une partie du gène est survenue uniquement chez l’enfant atteint d’autisme qui présente une absence totale de langage et un retard mental modéré. Dans un second cas, l’insertion d’une guanine est observée chez deux enfants atteints d’autisme ; elle modifie la phase de lecture de ce gène et entraîne la formation d’une protéine dépourvue de certains domaines d’interactions avec ses partenaires synaptiques (Figure 1). Ces deux enfants atteints d’autisme présentent un retard de développement du langage et un retard mental sévère. Cette mutation est absente chez le frère non atteint et chez les deux parents. L’analyse de la région chromosomique suggère une mosaïque germinale chez la mère (mutation présente dans les gamètes maternels). Dans une troisième famille, nous avons pu mettre en évidence une délétion du gène chez une fille atteinte d’autisme qui présente un retard mental et un retard de langage. Une duplication de la même région a été observée chez son frère qui présente un syndrome d’Asperger. Ces altérations, délétion et duplication, sont la conséquence d’une transmission déséquilibrée d’une translocation t(14 ;22)(p11.2;q13.33) équilibrée chez le père sain.
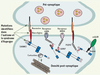 |
Figure 1. Schéma d’une synapse et des principales protéines synaptiques associées à l’autisme. La synapse glutamatergique du système nerveux central est caractérisée par une zone plus dense, appelée “la densité postsynaptique” (PSD : postsynaptic density). La PSD est caractérisée par un grand complexe protéique représenté en plusieurs classes : (1) récepteurs et canaux, (2) protéines du cytosquelette, (3) protéines d’échafaudage, (4) protéines d’adhésion cellulaires, (5) protéines G, (6) kinases et phosphatases. SAPAP : synapse-associated protein (SAP) 90/postsynaptic density (PSD)-95-associated protein ; AMPA : acide aminométhylphosphonique ; NMDA : N-méthyl-D-aspartate. |
Dans les différentes délétions observées, trois gènes au minimum sont affectés, et dans la translocation, le gène APPL2 qui code une protéine intervenant dans la prolifération cellulaire est également interrompu. Aussi, ces résultats génétiques témoignent, pour la première fois, du rôle majeur du gène SHANK3 dans le phénotype neurologique des délétions 22q13. Par ailleurs, ils mettent en évidence l’importance du dosage génique dans le développement du langage et de la communication sociale [16]. SHANK3 est une protéine d’échafaudage de la synapse qui permet, entre autres, de faire le lien entre les protéines membranaires et le cytosquelette d’actine. In vitro, l’expression de SHANK3 permet l’induction et le maintien des épines dendritiques, ainsi que le recrutement des récepteurs au glutamate à la membrane [17]. Notre analyse fonctionnelle montre que les mutations de SHANK3 altèrent la localisation synaptique de la protéine et potentiellement ses propriétés d’assemblage au niveau de la densité post-synaptique.
Conclusion
Les résultats obtenus sur les Neuroligines, SHANK3 et les Neurexines suggèrent fortement l’implication de la formation et la maturation des synapses dans l’étiologie de l’autisme [18]. Par ailleurs, les différents résultats génétiques et fonctionnels obtenus jusqu’à présent dans les cas d’autisme syndromique ont permis de suspecter l’implication de l’équilibre glutamate/GABA, et celui du maintien et de la formation des synapses. En effet, les résultats fonctionnels obtenus avec MeCP2 (impliqué dans le syndrome de Rett), les Neuroligines et les récepteurs GABA (duplications du chromosome 15), soulignent l’importance de ces gènes dans la balance excitation/inhibition et dans le fonctionnement des réseaux neuronaux. De plus, les mutations identifiées dans MeCP2, NLGN, FMR1 (le syndrome de l’X fragile), TSC1 et TSC2 (sclérose tubéreuse), et SHANK3 ont toutes un effet sur la morphologie et/ou le nombre des dendrites. Enfin, notre groupe a identifié récemment des défauts sévères de synthèse de la mélatonine chez certaines personnes avec autisme. Ainsi, une altération de l’effet modulateur de la mélatonine sur les réseaux neuronaux pourrait, dans certains cas, altérer les rythmes circadiens et augmenter la sévérité des atteintes neuronales chez les personnes avec autisme [19].
À la suite de l’identification de ces gènes, de nombreuses questions restent encore en suspens, qui concernent entre autres la spécificité des atteintes chez les patients et l’importance du dosage génique dans la mise en place de l’architecture synaptique. Pour répondre à ces nouvelles questions, de nouvelles technologies sont désormais possibles comme l’analyse comparée des résultats de la génétique et de l’imagerie cérébrale. Enfin, ces résultats génétiques confirment de nouveau que l’autisme est un syndrome génétiquement hétérogène. Aussi, il semble important que les moyens mis en œuvre pour les analyses cliniques (psychiatrie, neuropsychologie, génétique) et pour les analyses moléculaires (génétique et neurobiologie) soient renforcés afin de comprendre ce syndrome complexe qu’est l’autisme.
Remerciements
Les auteurs remercient les patients et leurs familles pour leur participation à ces études, l’Institut Pasteur, l’Inserm, l’Assistance Publique Hôpitaux de Paris, la Fondation France Telecom, Cure Autism Now, la Fondation de France, la Fondation Biomédicale de la Mairie de Paris, la Fondation pour la Recherche Médicale, EUSynapse European Commission, la Fondation NRJ pour leur soutien financier. Les auteurs remercient également le Centre d’Investigation Clinique de l’Hôpital Robert Debré, et la banque d’ADN de l’Hôpital Pitié-Salpêtrière.
Références
- Jamain S, Betancur C, Giros B, et al. La génétique de l’autisme. Med Sci (Paris) 2003; 19 : 1081–90. [Google Scholar]
- Persico AM, Bourgeron T. Searching for ways out of the autism maze : genetic, epigenetic and environmental clues. Trends Neurosci 2006; 29 : 349–58. [Google Scholar]
- Jamain S, Quach H, Betancur C, et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet 2003; 34 : 27–9. [Google Scholar]
- Chih B, Afridi SK, Clark L, Scheiffele P. Disorder-associated mutations lead to functional inactivation of neuroligins. Hum Mol Genet 2004; 13 : 1471–7. [Google Scholar]
- Tabuchi K, Blundell J, Etherton MR, et al. A Neuroligin-3 Mutation Implicated in Autism Increases Inhibitory Synaptic Transmission in Mice. Science 2007; 318 : 71–6. [Google Scholar]
- Varoqueaux F, Aramuni G, Rawson RL, et al. Neuroligins determine synapse maturation and function. Neuron 2006; 51 : 741–54. [Google Scholar]
- Talebizadeh Z, Lam DY, Theodoro MF, et al. Novel splice isoforms for NLGN3 and NLGN4 with possible implications in autism. J Med Genet 2006; 43 : e21. [Google Scholar]
- Laumonnier F, Bonnet-Brilhault F, Gomot M, et al. X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am J Hum Genet 2004; 74 : 552–7. [Google Scholar]
- Yan J, Oliveira G, Coutinho A, et al. Analysis of the neuroligin 3 and 4 genes in autism and other neuropsychiatric patients. Mol Psychiatry 2005; 10 : 329–32. [Google Scholar]
- Vincent JB, Kolozsvari D, Roberts WS, et al. Mutation screening of X-chromosomal neuroligin genes : no mutations in 196 autism probands. Am J Med Genet B Neuropsychiatr Genet 2004; 129 : 82–4. [Google Scholar]
- Gauthier J, Bonnel A, St-Onge J, et al. NLGN3/NLGN4 gene mutations are not responsible for autism in the Quebec population. Am J Med Genet B Neuropsychiatr Genet 2005; 132 : 74–5. [Google Scholar]
- Blasi F, Bacchelli E, Pesaresi G, et al. Absence of coding mutations in the X-linked genes neuroligin 3 and neuroligin 4 in individuals with autism from the IMGSAC collection. Am J Med Genet B Neuropsychiatr Genet 2006; 141 : 220–1. [Google Scholar]
- Szatmari P, Paterson AD, Zwaigenbaum L, et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet 2007; 39 : 319–28. [Google Scholar]
- Manning MA, Cassidy SB, Clericuzio C, et al. Terminal 22q deletion syndrome : a newly recognized cause of speech and language disability in the autism spectrum. Pediatrics 2004; 114 : 451–7. [Google Scholar]
- Bonaglia MC, Giorda R, Borgatti R, et al. Disruption of the ProSAP2 gene in a t(12;22)(q24.1;q13.3) is associated with the 22q13.3 deletion syndrome. Am J Hum Genet 2001; 69 : 261–8. [Google Scholar]
- Durand CM, Betancur C, Boeckers TM, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet 2007; 39 : 25–7. [Google Scholar]
- Roussignol G, Ango F, Romorini S, et al. Shank expression is sufficient to induce functional dendritic spine synapses in aspiny neurons. J Neurosci 2005; 25 : 3560–70. [Google Scholar]
- Belmonte MK, Bourgeron T. Fragile X syndrome and autism at the intersection of genetic and neural networks. Nat Neurosci 2006; 9 : 1221–5. [Google Scholar]
- Melke J, Goubran Botros H, et al. Abnormal melatonin synthesis in autism spectrum disorders. Mol Psychiatry 2007 15 mai online. [Google Scholar]
© 2008 médecine/sciences - Inserm / SRMS
Liste des tableaux
Bilan des études des gènes NLGN3 et NLGN4 dans l’autisme et le retard mental. Asp : syndrome d’Asperger ; RM : retard mental ; TSA : troubles du spectre autistique.
Liste des figures
|
Figure 1. Schéma d’une synapse et des principales protéines synaptiques associées à l’autisme. La synapse glutamatergique du système nerveux central est caractérisée par une zone plus dense, appelée “la densité postsynaptique” (PSD : postsynaptic density). La PSD est caractérisée par un grand complexe protéique représenté en plusieurs classes : (1) récepteurs et canaux, (2) protéines du cytosquelette, (3) protéines d’échafaudage, (4) protéines d’adhésion cellulaires, (5) protéines G, (6) kinases et phosphatases. SAPAP : synapse-associated protein (SAP) 90/postsynaptic density (PSD)-95-associated protein ; AMPA : acide aminométhylphosphonique ; NMDA : N-méthyl-D-aspartate. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.