")
")
| Issue |
Med Sci (Paris)
Volume 23, Number 6-7, Juin-Juillet 2007
|
|
|---|---|---|
| Page(s) | 565 - 567 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/20072367565 | |
| Published online | 15 juin 2007 | |
Réactivation de p53 dans les tumeurs : une stratégie antitumorale prometteuse
The activation of p53 in tumors: a promising strategy against cancer
Université Pierre et Marie Curie-Paris 6, Institut Curie, Centre de Recherche, Génétique de la Suppression Tumorale, UMR 7147 IC/UPMC/CNRS, 26, rue d’Ulm, 75248 Paris Cedex 05, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
La mutation du gène de p53 dans la moitié des cancers humains, et l’altération de protéines régulant p53 dans l’autre moitié des cancers, témoignent du rôle essentiel de p53 dans la suppression tumorale. Ce facteur de transcription agit comme un gardien du génome : en réponse à divers stress cellulaires, comme l’activation d’oncogènes ou la présence de cassures de l’ADN, p53 est stabilisée et activée, et induit la transcription de multiples gènes. Cette réponse transcriptionnelle peut conduire à un arrêt du cycle cellulaire réversible, qui permet la réparation de l’ADN avant de poursuivre le cycle, ou induire des réponses irréversibles, comme un arrêt de prolifération permanent (sénescence) ou un programme de mort cellulaire (apoptose). Les facteurs conditionnant l’initiation d’un type de réponse plutôt qu’un autre ne sont pas totalement compris. De fait, la régulation des réponses transcriptionnelles de p53 apparaît particulièrement complexe, puisque environ 160 protéines influeraient sur la stabilité ou l’activité de cette protéine [1], et que p53 contrôle l’expression de plusieurs centaines de gènes [2]. Malgré sa complexité, l’importance de cette voie de régulation dans la suppression tumorale a suscité le développement de diverses stratégies visant à réactiver p53 dans les cancers, dans le but de ralentir la progression tumorale, voire de résorber les tumeurs. Ces stratégies reposent sur une modulation des protéines qui paraissent primordiales dans cette voie de régulation (Figure 1).
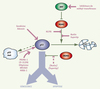 |
Figure 1. Stratégies d’activation de p53 dans les tumeurs. À gauche : dans la moitié des tumeurs, p53 est absente ou mutée (p53 mut). La réintroduction de p53 sauvage par thérapie génique (Gendicine, Advexin) ou la restauration d’une conformation sauvage de p53 par différentes molécules (PRIMA-1, etc.) peuvent être envisagées. À droite : dans l’autre moitié des cancers, une p53 sauvage est présente mais sa stabilité ou son activité est altérée, le plus souvent à cause de la surexpression des oncogènes MDM2 ou MDM4, ou de l’inactivation du suppresseur de tumeurs ARF. Dans ces cas, des inhibiteurs de méthyl-transférases peuvent réactiver ARF, HLI98 inhiber l’activité ubiquitine-ligase de MDM2, et les Nutlins ou Supertips être efficaces pour bloquer les interactions MDM2-p53, mais peu efficaces envers MDM4. Toutes ces approches visent à restaurer la fonction de p53 sauvage et conduire la cellule tumorale vers la sénescence ou l’apoptose. |
Un premier type de stratégie concerne la moitié des cancers qui n’exprime pas p53 ou qui exprime une p53 mutée. Dans ces tumeurs, une approche possible est celle de la thérapie génique, c’est-à-dire la réintroduction d’un gène p53 sauvage : un adénovirus recombinant de type 5 porteur du gène p53 est actuellement commercialisé en Chine sous le nom de Gendicine, et un produit similaire, l’Advexin, fait l’objet de tests cliniques en phase III aux États-Unis. Une utilisation combinée de ce type de vecteur avec les approches classiques de radiothérapie ou de chimiothérapie semble donner de bons résultats pour les cancers épidermoïdes de la tête et du cou, mais le traitement est coûteux, son efficacité thérapeutique pour d’autres types de cancers n’est pas établie, et la dangerosité potentielle des vecteurs adénoviraux reste préoccupante [3]. Une autre approche consiste à tenter de corriger la conformation de la protéine p53 mutante. En effet, un grand nombre de mutations de p53 perturbent sa structure, et réduisent ainsi son efficacité à lier l’ADN et activer la transcription de ses gènes cibles. Plusieurs molécules corrigeant la structure de p53 mutantes ont été identifiées. Parmi elles, la molécule PRIMA-1, en favorisant la conformation sauvage de certaines p53 mutantes, induit une réponse apoptotique et permet de limiter la croissance de tumeurs humaines greffées dans des souris SCID (severe combined immunodeficient). Cependant, chacune de ces molécules ne peut corriger qu’une fraction des p53 mutantes, et certaines molécules semblent également affecter la conformation d’autres protéines [4].
Dans les cellules cancéreuses exprimant une p53 sauvage, divers mécanismes sont responsables de l’inhibition de la voie de régulation de p53. Ainsi, dans 10 % des cancers, l’inactivation de p53 est la conséquence d’une amplification du gène codant MDM2, un inhibiteur spécifique de p53 [1]. MDM2 exerce principalement son activité inhibitirice en ubiquitinylant p53, ce qui conduit à la dégradation de p53 par le protéasome (Figure 1). La caractérisation par cristallographie du mode d’interaction de ces 2 protéines a montré le rôle essentiel de 3 acides aminés de p53 qui se logent dans une poche hydrophobe à la surface de MDM2. Ces données ont permis d’identifier des peptides (Supertip) ou des petites molécules (Nutlins) capables de se loger dans la poche hydrophobe de MDM2, permettant ainsi à p53 d’échapper à l’action inhibitrice de MDM2. Il a été montré que les Nutlins1 inhibent la croissance de certaines cellules tumorales xénogreffées dans des souris nude [5]. Une autre approche a été de chercher des molécules inhibant l’activité ubiquitine ligase de MDM2, ce qui a conduit à identifier le composé HLI98, qui n’a cependant pas été testé in vivo [4]. En réponse à un stress oncogénique (surexpression de Myc, activation de Ras…), ARF inhibe MDM2, et agit donc comme l’autre suppresseur de tumeurs dans cette voie de régulation (Figure 1). ARF est l’un des 2 transcrits du locus CDKN2A, dont la délétion est observée dans certaines tumeurs, mais dont l’expression est plus fréquemment abolie par la méthylation de son promoteur [6]. Des inhibiteurs de méthyl transférases, actuellement en essais cliniques, pourraient entraîner la réexpression de ARF dans certaines tumeurs et réactiver la voie p53 (Figure 1). Cela dit, dans les tumeurs n’exprimant pas ARF, p53 pourrait également être réactivée en inhibant MDM2 à l’aide de Nutlins. Un autre acteur majeur de cette voie de régulation est MDM4 (aussi connu sous le nom de MDMX). MDM4 est un inhibiteur spécifique de p53 qui est amplifié ou surexprimé dans 10 à 20 % des cancers [1], et jusqu’à 65 % des cas de rétinoblastomes [7]. MDM4 partage des similitudes structurales avec MDM2. Cependant, il ne contrôle pas la stabilité de p53, mais son activité transcriptionnelle [8, 9] (Figure 1). MDM4 se lie à p53 grâce à une poche hydrophobe de structure voisine, mais pas identique, à celle présente à la surface de MDM2. Il en résulte que des molécules antagonistes de l’interaction entre MDM2 et p53, comme Supertip 12/1 ou Nutlin 3, sont des antagonistes peu efficaces de l’interaction entre MDM4 et p53 [1, 7] (Figure 1). La recherche d’antagonistes de MDM4 plus efficaces est un objectif clinique important, puisque l’inhibition spécifique de MDM4 dans les cellules tumorales limite la croissance des tumeurs xénogreffées [7, 8]. Par ailleurs, une meilleure compréhension de la voie de régulation p53 devrait suggérer d’autres stratégies : ainsi les protéines HAUSP et Daxx, récemment identifiées comme régulatrices de la stabilité de p53, MDM2 et MDM4, pourraient aussi devenir des cibles thérapeutiques intéressantes [10].
L’ensemble de ces résultats indique que la restauration de l’activation de p53 dans les cellules tumorales est une stratégie particulièrement prometteuse pour le traitement d’un grand nombre de cancers. Néanmoins, deux points majeurs restaient à éclaircir : (1) celui de l’efficacité de ces stratégies. En effet, dans la plupart de ces études, le ralentissement de la croissance tumorale a été testé in vivo sur des tumeurs xénogreffées [4, 5, 7, 8], mais n’a pas été évalué dans le cas de tumeurs préexistantes in situ. La seule exception était celle des tests avec les adénovirus recombinants, mais ceux-ci se montraient peu efficaces dans un grand nombre de cas de cancers [3] ; (2) celui de l’index thérapeutique de ces approches : en effet, l’administration orale d’un médicament activant la voie p53 pourrait conduire à une activation de p53 dans les cellules tumorales, mais peut-être aussi dans les cellules normales, et se révéler toxique. Certaines données suggéraient une sensibilité accrue des cellules tumorales à la Nutlin du fait d’une altération de leur transcriptome [11], mais d’autres montraient que l’inactivation totale de MDM2 dans des cellules normales est rapidement létale chez la souris [12].
Trois nouveaux modèles murins apportent maintenant des informations cruciales sur ces points et révèlent également des informations inattendues. Ces trois modèles utilisent des stratégies génétiques sophistiquées pour réactiver p53 dans des tumeurs in situ. Ainsi, Ventura et al. ont combiné le système de recombinaison Cre-Lox et le système de translocation ERTAM pour créer des souris dans lesquelles p53 n’est exprimée qu’après addition de tamoxifène [13]. Dans ces souris, des signaux d’arrêt de la transcription et de la traduction ont été introduits en amont des séquences codantes de p53, empêchant la production de la protéine. Ces stop sont flanqués par des sites Lox, sites de reconnaissance spécifiques de la recombinase Cre. Par ailleurs, les souris expriment, dans tous leurs tissus, une protéine de fusion entre la recombinase Cre et le récepteur des œstrogènes (ERTAM). Cette protéine Cre-ERTAM est cytoplasmique, mais l’addition de tamoxifène provoque sa translocation dans le noyau, où elle reconnaît les sites Lox, ce qui conduit à l’excision des signaux stop et permet l’expression de p53. Ainsi, le phénotype de ces souris est équivalent à celui de souris déficientes en p53, mais devient équivalent à celui de souris sauvages après injection de tamoxifène. Comme les souris déficientes en p53, ces souris développent des lymphomes et des sarcomes. L’injection intrapéritonéale de tamoxifène conduit alors à la régression de ces deux types de tumeurs in situ. Le tamoxifène conduit également à une expression du gène de p53 dans les cellules non tumorales, mais sans effet toxique, ce qui démontre l’excellent index thérapeutique de ce type d’approche. Les résultats suggèrent que dans les cellules de lymphomes et de sarcomes, une activation d’oncogènes conduit à l’induction de ARF, ce qui sensibilise les cellules tumorales à l’expression de p53.
Par ailleurs, cette étude révèle que la réactivation de p53 conduit à une régression rapide des lymphomes, due à l’activation d’un programme apoptotique. En revanche, la réactivation de p53 dans les cellules de sarcomes conduit à leur sénescence, et la régression de ces tumeurs est plus lente. Comment se produit cette régression ? L’étude de Xue et al. suggère que la réponse immunitaire est impliquée [14]. Xue et al. ont isolé des cellules progénitrices de foie, qu’ils ont infectées avec des rétrovirus permettant l’expression constitutive de l’oncogène activé Ras et l’expression conditionnelle d’un short hairpin ARN (shARN) spécifique de p53. Dans ces cellules, p53 est inactivée par l’interférence par l’ARN due au shARN, mais l’ajout de doxycycline réprime cet ARN et rétablit l’expression de p53. L’injection de ces cellules dans le foie conduit à l’apparition rapide d’hépatocarcinomes, qui régressent après administration de doxycycline. La régression de ces tumeurs serait due à la sénescence des cellules tumorales, puis à une réaction inflammatoire qui détruit ces cellules. La régression de ces tumeurs est d’ailleurs bien moindre lorsque les mêmes expériences sont réalisées dans des souris NOD/SCID, dont le système immunitaire est très affaibli [14]. Enfin, dans une troisième étude, Martins et al. ont étudié les conséquences de la surexpression de Myc dans des souris Eµ-Myc p53+/KI, où l’allèle p53KI exprime une protéine de fusion p53-ERTAM [15]. Comme les souris Eµ-Myc p53+/-, ces souris développent rapidement des lymphomes en perdant l’allèle sauvage de p53. Cependant, l’ajout de tamoxifène dans ces lymphomes permet à la p53-ERTAM d’activer ses gènes cibles et d’induire une apoptose, et donc la régression rapide des tumeurs. Mais l’émergence de tumeurs résistantes est fréquente dans ce système, et résulte de la perte de l’allèle p53-ERTAM ou de ARF [15].
Ensemble, ces études montrent que la réactivation de p53 dans les tumeurs peut conduire à leur régression par des mécanimes différents selon les tissus, mais que des stratégies doivent être mises au point pour optimiser la réponse p53 tout en limitant l’apparition de tumeurs résistantes. Selon toute vraisemblance, la combinaison de différentes approches (chimiothérapie classique, activation de la voie p53, activation de la réponse immunitaire) devrait permettre de limiter l’émergence de cellules résistantes.
Le terme nutlin vient du nom du site de la firme Hoffmann-La Roche aux États-Unis - Nutley - dans le New Jersey.
Références
- Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer 2006; 6 : 909–23. [Google Scholar]
- Vousden KH. Outcomes of p53 activation: spoilt for choice. J Cell Sci 2006; 119 : 5015–20. [Google Scholar]
- Jia H. Controversial Chinese gene-therapy drug entering unfamiliar territory. Nat Rev Drug Discov 2006; 5 : 269–70. [Google Scholar]
- Wiman KG. Strategies for therapeutic targeting of the p53 pathway in cancer. Cell Death Differ 2006; 13 : 921–6. [Google Scholar]
- Vassilev LT. MDM2 inhibitors for cancer therapy. Trends Mol Med 2007; 13 : 23–31. [Google Scholar]
- Sherr CJ. Divorcing ARF and p53: an unsettled case. Nat Rev Cancer 2006; 6 : 663–73. [Google Scholar]
- Laurie NA, Donovan SL, Shih CS, et al. Inactivation of the p53 pathway in retinoblastoma. Nature 2006; 444 : 61–6. [Google Scholar]
- Toledo F, Krummel KA, Lee CJ, et al. A mouse p53 mutant lacking the proline rich domain rescues Mdm4 deficiency and provides insight into the Mdm2-Mdm4-p53 regulatory network. Cancer Cell 2006; 9 : 273–85. [Google Scholar]
- Francoz S, Froment P, Bogaerts S, et al. Mdm4 and Mdm2 cooperate to inhibit p53 activity in proliferating and quiescent cells in vivo. Proc Natl Acad Sci USA 2006; 103 : 3232–7. [Google Scholar]
- Ronai Z. Balancing Mdm2: a Daxx-HAUSP matter. Nat Cell Biol 2006; 8 : 790–1. [Google Scholar]
- Brummelkamp TR, Fabius AW, Mullenders J, et al. An shRNA barcode screen provides insight into cancer cell vulnerability to MDM2 inhibitors. Nat Chem Biol 2006; 2 : 202–6. [Google Scholar]
- Ringshausen I, O’Shea C, Finch AJ, et al. Mdm2 is critically and continuously required to suppress lethal p53 activity in vivo. Cancer Cell 2006; 10 : 501–14. [Google Scholar]
- Ventura A, Kirsch DG, McLaughlin ME, et al. Restoration of p53 function leads to tumour regression in vivo. Nature 2007; 445 : 661–5. [Google Scholar]
- Xue W, Zender L, Miething C, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007; 445 : 656–60. [Google Scholar]
- Martins CP, Brown-Swigart L, Evan GI. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell 2006; 127 : 1323–34. [Google Scholar]
© 2007 médecine/sciences - Inserm / SRMS
Liste des figures
|
Figure 1. Stratégies d’activation de p53 dans les tumeurs. À gauche : dans la moitié des tumeurs, p53 est absente ou mutée (p53 mut). La réintroduction de p53 sauvage par thérapie génique (Gendicine, Advexin) ou la restauration d’une conformation sauvage de p53 par différentes molécules (PRIMA-1, etc.) peuvent être envisagées. À droite : dans l’autre moitié des cancers, une p53 sauvage est présente mais sa stabilité ou son activité est altérée, le plus souvent à cause de la surexpression des oncogènes MDM2 ou MDM4, ou de l’inactivation du suppresseur de tumeurs ARF. Dans ces cas, des inhibiteurs de méthyl-transférases peuvent réactiver ARF, HLI98 inhiber l’activité ubiquitine-ligase de MDM2, et les Nutlins ou Supertips être efficaces pour bloquer les interactions MDM2-p53, mais peu efficaces envers MDM4. Toutes ces approches visent à restaurer la fonction de p53 sauvage et conduire la cellule tumorale vers la sénescence ou l’apoptose. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.