")
")
| Issue |
Med Sci (Paris)
Volume 23, Number 3, Mars 2007
|
|
|---|---|---|
| Page(s) | 261 - 262 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/2007233261 | |
| Published online | 15 mars 2007 | |
Une nouvelle forme d’ataxie récessive causée par des mutations du gène SYNE-1
Mutations in SYNE-1 lead to a newly discovered form of autosomal recessive cerebellar ataxia
1
Centre de recherche sur les maladies du cerveau, Université de Montréal, CHUM (Notre-Dame), Pavillon J.A. de Sève, 1560, Sherbrooke Est, Montréal, Québec, H2L 4M1 Canada
2
Faculté de Médecine, Université Laval, Département des sciencesneurologiques, CHAUQ (Enfant-Jésus), 1401, 18e Rue, Québec, G1J 1Z4 Canada
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
L’ataxie se traduit par des difficultés, pour le sujet atteint, de se maintenir en équilibre ; cette affection peut être le résultat de pathologies héréditaires ou acquises. Les ataxies héréditaires sont en général dominantes ou récessives ; elles ont certes comme caractéristique commune une perturbation de l’équilibre, cependant elles sont souvent associées à d’autres affections neurologiques : dégénérescence des faisceaux pyramidaux, neuropathie, déclin cognitif, rétinopathie ou encore signes extra-pyramidaux. Parmi les ataxies héréditaires récessives dont le gène a été identifié à ce jour, on relève principalement l’ataxie de Friedreich, l’ataxie avec télangiectasie, l’ataxie récessive spastique de Charlevoix-Saguenay (ARSACS) et l’ataxie avec déficience en vitamine E.
Entre 1996 et 2006, nous avons caractérisé plusieurs familles canadiennes-françaises originaires de la Beauce et du Bas-St-Laurent (Québec, Canada) dont plusieurs membres présentaient une ataxie d’apparition tardive et de transmission récessive très homogène sur le plan clinique [1, 2]. Nous avons nommé cette nouvelle forme d’ataxie autosomal recessive cerebellar ataxia type 1 (ARCA-1), également connue sous le nom d’Ataxie Récessive de la Beauce (ARB). La population canadienne-française étant issue d’un nombre limité de fondateurs dont les descendants ont été relativement isolés au cours des générations, on y décèle un bon nombre de maladies héréditaires pourtant rares au sein d’autres populations. L’ARCA-1 s’ajoute donc aux maladies héréditaires neurologiques propres à cette population comme, par exemple, l’ARSACS [3] ou la neuropathie sensitivomotrice héréditaire avec agénésie du corps calleux (HMSN/ACC) [4].
Afin de mieux définir l’ARCA-1, nous avons mené une étude phénotypique détaillée de 26 familles d’origine canadienne-française dont 53 membres présentaient une ataxie cérébelleuse pure caractérisée seulement par un trouble de la marche et un trouble de l’élocution à l’exclusion d’autres signes d’atteinte neurologique significative [5]. Ainsi, l’ARCA-1 se définit principalement par de la dysarthrie ou de l’ataxie cérébelleuse débutant à l’âge adulte (âge de début moyen 30,4 années ; intervalle de 17-46 ans). Cette nouvelle forme d’ataxie est associée à des anomalies mineures des mouvements extra-oculaires (réduction de la vitesse des saccades), une incoordination des membres ou encore des réflexes ostéotendineux parfois vifs aux membres inférieurs. Sur le plan des examens para-cliniques, l’imagerie par résonance magnétique révèle une atrophie cérébelleuse diffuse sans atteinte du tronc cérébral chez l’ensemble des sujets atteints après quelques années seulement d’évolution de la maladie (Figure 1).
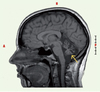 |
Figure 1. Imagerie par résonance magnétique d’une patiente de 29 ans atteinte de l’ARCA-1. Cette coupe sagittale T1 montre une atrophie cérébelleuse diffuse. |
Afin de déterminer le gène responsable de l’ARCA-1, nous avons d’abord fait une étude génomique complète avec 388 marqueurs microsatellites à des intervalles approximatifs de 10cM, en utilisant les ADN de 20 patients et de 6 membres non-atteints de leurs familles [5]. Cette étude de localisation montrait un marqueur de microsatellite (D6S476) avec un score LOD supérieur à trois. Par la suite, nous avons génotypé 21 marqueurs additionnels voisins de D6S476, ce qui nous a permis de définir un intervalle de 0,5 Mb sur le chromosome 6q entre les marqueurs D6S420 et GATA186B06. Le seul gène inclus dans cet intervalle, SYNE-1 (synaptic nuclear envelope protein 1), s’étend sur plus de 0,5 Mb et est constitué de 147 exons qui codent un ARN messager de 27 652 kb et une protéine de 8 797 acides aminés. Afin de découvrir les mutations responsables de l’ARCA-1, nous avons établi la séquence complète, chez tous les patients ARCA-1 identifiés, des exons du gène SYNE-1 incluant les jonctions introns/exons. Cette opération nous a permis d’identifier 5 mutations causales au sein de notre population (Figure 2). Chaque mutation repose sur son propre haplotype, dont deux sont nettement plus fréquents, ce qui confirme l’effet fondateur que nous avions anticipé au sein de cette population relativement homogène d’un point de vue ancestral.
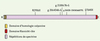 |
Figure 2. Structure de la protéine SYNE-1 avec les mutations identifiées à ce jour. C : carboxy-terminal ; N : amino-terminal. |
Avant notre découverte, la seule fonction alors connue du gène SYNE-1 était celle de l’ancrage des noyaux des fibres musculaires des jonctions neuromusculaires [6]. Afin de vérifier si les patients atteints de l’ARCA-1 présentaient des anomalies dans la migration des noyaux des fibres musculaires des jonctions neuromusculaires, nous avons étudié un échantillon de muscle d’un sujet atteint et d’un sujet témoin, révélant ainsi que les noyaux musculaires synaptiques se situaient directement sous les synapses dans seulement 40 % des jonctions neuromusculaires étudiées chez le sujet atteint contre 80 % des jonctions du sujet témoin [5]. Bien qu’elle ne soit pas associée à une atteinte clinique, cette propriété des fibres musculaires coïncide avec les connaissances actuelles sur SYNE-1. Cependant, notre étude a également mis en lumière une fonctionnalité nouvelle de SYNE-1 dans le cervelet compte tenu de l’atrophie observée chez tous les patients ARCA-1. Nous avons donc étudié l’expression et la localisation de SYNE-1 dans le système nerveux chez la souris, ce qui nous a permis de démontrer que ce gène était exprimé à un haut degré dans le cervelet (cellules de Purkinje) et dans le tronc cérébral (noyaux olivaires) [5]. La protéine codée par SYNE-1 est constituée des portions suivantes : une paire de domaines homologues au domaine transmembranaire calponine, de nombreuses répétitions de spectrine et un domaine klarsicht1 en carboxy-terminal. Le domaine spectrine jouerait un rôle dans la formation et la maintenance des structures cellulaires. Comme les mutations décelées chez les patients ARCA-1 sont toutes situées dans le domaine spectrine, on peut émettre l’hypothèse que les mutations repérées chez les patients atteints de l’ARCA-1 affectent la capacité de SYNE-1 de se lier à l’actine ou à d’autres ligands, ce qui semble avoir une répercussion significative sur la structure nucléaire des cellules de Purkinje.
Ainsi, nous avons décrit une nouvelle maladie : il s’agit de la plus fréquente ataxie héréditaire au Québec après l’ARSACS et l’ataxie de Friedreich. Nous avons révélé pour la première fois que le gène SYNE-1 pouvait être associé à une maladie chez l’humain ; de plus, nous avons mis en lumière une fonction nouvelle de ce gène dans le cervelet. Cela nous amène à définir une nouvelle catégorie d’ataxies héréditaires - celles reliées au domaine spectrine - qui comprend désormais ARCA-1, SCA-5 [7] et 15q-ADCA [8], toutes trois caractérisées par une dégénérescence cérébelleuse progressive.
Le domaine KASH (Klarsicht, ANC-1, Syne homology) est en position carboxy-terminale de certaines protéines retrouvées chez la drosophile (Klarsicht et MSP-300), C. elegans (ANC-1), et les mammifères (Syne-1 et Syne-2). Ce domaine transmembranaire serait impliqué dans l’ acheminement de la protéine vers la membrane nucléaire cellulaire.
Références
- Dupré N, Bouchard JP, Verreault S, et al. Recessive ataxia of the Beauce, a new form of hereditary ataxia of pure cerebellar type. Neurology 2002; 58 :A35. [Google Scholar]
- Dupré N, Gros-Louis F, Verreault S, et al. Recessive ataxia of the Beauce, a new form of hereditary ataxia, maps to chromosome 6. Neurology 2006; 66 : A274. [Google Scholar]
- Dupré N, Bouchard JP, Brais B, Rouleau GA. Hereditary ataxia, spastic paraparesis and neuropathy in the French-Canadian population. Can J Neurol Sci 2006; 33 : 149–57. [Google Scholar]
- Howard HC, Dupré N, Mathieu J, et al. Severe neuropathy with agenesis of the corpus callosum. Med Sci (Paris) 2003; 19 : 414–6. [Google Scholar]
- Gros-Louis F, Dupré N, Dion P, et al. Mutations in SYNE1 lead to a newly discovered form of autosomal recessive cerebellar ataxia. Nat Genet 2007; 39 : 80–85 [Google Scholar]
- Grady RM, Starr DA, Ackerman GL, et al. Syne proteins anchor muscle nuclei at the neuromuscular junction. Proc Natl Acad Sci USA 2005; 102 : 4359–64. [Google Scholar]
- Ikeda Y, Dick KA, Weatherspoon MR, et al. Spectrin mutations cause spinocerebellar ataxia type 5. Nat Genet 2006; 38 : 184–90. [Google Scholar]
- Ishikawa K, Toru S, Tsunemi T, et al. An autosomal dominant cerebellar ataxia linked to chromosome 16q22.1 is associated with a single-nucleotide substitution in the 5’ untranslated region of the gene encoding a protein with spectrin repeat and Rho guanine-nucleotide exchange-factor domains. Am J Hum Genet 2005; 77 : 280–96. [Google Scholar]
© 2007 médecine/sciences - Inserm / SRMS
Liste des figures
|
Figure 1. Imagerie par résonance magnétique d’une patiente de 29 ans atteinte de l’ARCA-1. Cette coupe sagittale T1 montre une atrophie cérébelleuse diffuse. |
| Dans le texte | |
|
Figure 2. Structure de la protéine SYNE-1 avec les mutations identifiées à ce jour. C : carboxy-terminal ; N : amino-terminal. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.