")
")
| Issue |
Med Sci (Paris)
Volume 23, Number 2, Février 2007
|
|
|---|---|---|
| Page(s) | 187 - 192 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/2007232187 | |
| Published online | 15 février 2007 | |
Pollution atmosphérique et maladies respiratoires allergiques
Air pollution and allergic airway diseases
1
Inserm U700 et Service de Physiologie-Explorations fonctionnelles
2
Inserm U700 et Service de Pneumologie A, Université Paris 7
3
Hôpital Bichat-Claude-Bernard, AP-HP, 46, rue Henri Huchard, 75018 Paris, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Les particules diesel (PDi) exercent une activité adjuvante sur la production d’IgE (animal sensibilisé à un allergène et injection simultanée de l’allergène et des PDi), et cela quelle que soit la voie d’administration utilisée. Chez l’homme, l’instillation nasale de PDi chez des volontaires sains provoque une augmentation de la production de cytokines pro-Th2, caractéristiques de la réponse allergique. À partir des études réalisées chez l’homme et chez l’animal, on peut donc proposer une hypothèse expliquant la recrudescence des pathologies respiratoires, et notamment des crises d’asthme, observée lors des épisodes de pollution particulaire.
Abstract
In the last decades, many studies have shown an increase in the prevalence of allergic rhinitis and asthma mainly in urban communities, especially in industrialized countries. Airborne pollutants such as diesel exhaust particles, ozone, nitrogen dioxide and sulphur dioxide have been implicated in the initiation and exacerbation of allergic airway diseases. Epidemiologic studies have shown clear associations between air pollution and allergic diseases, in vivo and in vitro studies have provided biologic link and potential molecular mechanisms. Particulate and gaseous pollutants can act both on the upper and lower airways to initiate and exacerbate cellular inflammation through interaction with the innate immune system. As a consequence, increased non-specific airway hyper-responsiveness and airway resistance have been observed in man. Diesel exhaust particles can both induce and exacerbate in vivo allergic responses. They can also modify the immune system’s handling of the allergen. The effects of gaseous pollutants on immune responses to allergens are not fully understood. We review the different mechanisms involved in the enhancement of allergic inflammation by urban air pollutants, including effects on cytokine and chemokine production, as well as activation of different immune cells. We discuss the hypothesis that pollutants’ effects on the immune system involve hierarchical oxidative stress. Susceptibility genes to air pollution inducing allergic diseases are also discussed.
© 2007 médecine/sciences - Inserm / SRMS
L’incidence des maladies allergiques a augmenté dans la plupart des pays industrialisés. L’exposition chronique aux particules de la pollution atmosphérique, produites en grande partie par la circulation automobile, est l’un des facteurs incriminés dans l’augmentation de la prévalence des maladies allergiques respiratoires [1, 2]. Les études épidémiologiques conduites dans différents pays ont démontré une forte association entre le niveau des émissions liées au trafic des véhicules à moteur et l’augmentation des symptômes d’asthme ou de rhinite allergique.
L’épidémiologie permet de mettre en évidence des associations statistiques entre les concentrations en un ou plusieurs polluants atmosphériques et des effets comme la fréquence des visites aux urgences ou celle des admissions à l’hôpital pour crise d’asthme. Cependant, ce type d’étude ne prouve pas de lien de causalité. Les modèles animaux et les études in vitro, quant à eux, apportent des informations sur les mécanismes de réponse lors de l’exposition aux polluants.
Enfin, les études d’exposition chez l’homme permettent d’évaluer les effets d’environnements contrôlés chez les populations à haut risque (comme les sujets asthmatiques), tout en évitant les facteurs confondants souvent présents en épidémiologie. Ces travaux ont d’abord testé les modifications à court terme de la fonction respiratoire et explorent actuellement les interactions et possibles synergies de multiples polluants entre eux, ou de polluants avec les allergènes.
Classification des polluants atmosphériques
Les polluants atmosphériques peuvent être classés en fonction de leur source de production, leur composition chimique, leur taille et leur mode de diffusion dans l’environnement. Les polluants directement émis dans l’atmosphère sont dits primaires, tandis que les polluants formés à partir de réactions chimiques avec d’autres polluants ou des gaz atmosphériques sont dits secondaires (Encadré). Les polluants particulaires en suspension, produits en grande partie par la circulation automobile, notamment les véhicules diesel, sont classés en trois catégories selon leur diamètre aérodynamique moyen (Encadré). Les particules ultrafines de diamètre aérodynamique moyen inférieur à 0,1 µm sont les plus abondantes, et leurs effets sur la santé sont peu connus [1]. Les polluants atmosphériques les plus abondants en milieu urbain sont les particules diesel, les oxydes d’azote et l’ozone.
Effets des polluants et des interactions allergènes-polluants
Particules diesel
Les particules diesel constituent la majeure partie de la pollution particulaire urbaine. Elles proviennent des moteurs de type diesel, dont la combustion produit des oxydes d’azote, des précurseurs d’ozone et des suies sous forme de microsphères de carbone agrégées les unes aux autres, avec un diamètre aérodynamique moyen de 100 nm. Grâce à leur grande surface d’exposition, les particules diesel permettent à de multiples molécules d’y être adsorbées : produits de la combustion incomplète, comme les hydrocarbures aromatiques polycycliques (HAP), les sulfates et les métaux lourds, mais aussi autres types d’aérocontaminants, et notamment les pollens. L’absence de biomarqueur fiable du niveau d’exposition aux particules diesel (taille des particules, teneur en HAP…) explique que la plupart des travaux épidémiologiques comparent des populations plus ou moins proches de zones de fort trafic routier. Plusieurs études ont montré que la proximité de ce trafic était un facteur de risque associé à l’asthme et à l’atopie [2–5].

Les effets spécifiques des particules diesel sur les maladies allergiques respiratoires ont été analysés chez l’animal, ainsi qu’in vitro et in vivo chez l’homme. Les principaux résultats suggèrent un effet adjuvant des particules diesel sur le développement et l’intensité des réponses inflammatoires allergiques.
Dans les modèles animaux, l’exposition aux particules diesel induit une augmentation de l’hyperréactivité bronchique et de l’inflammation des voies aériennes d’origine allergique [2, 6] (Tableau I). Lors d’une exposition combinée avec des allergènes, les particules diesel agissent comme des adjuvants en augmentant les réponses humorales IgE dépendantes, en induisant l’expression de cytokines de type TH2 [2, 7] et en majorant l’hyperréactivité bronchique [8]. Chez le cobaye exposé aux particules diesel, l’administration nasale répétée d’allergènes augmente les éternuements, les sécrétions nasales et l’infiltration muqueuse par les polynucléaires éosinophiles [9].
Effets adjuvants des particules diesel sur la réponse inflammatoire allergique. GM-CSF : granulocyte macrophage colony stimulating factor ; TH2 : T helper de type 2 ; RANTES : regulation upon activation normal T cell expressed and secreted ; MCP-3 : macrophage chemoattractant protein-3 ; MIP-1α : macrophage inflammatory protein-1α ; KLH : keyhole limpet hemocyanin.
Les études in vitro montrent que les particules diesel peuvent agir sur de nombreux types cellulaires [2, 10] (Figure 1). Elles favorisent la production de médiateurs pro-inflammatoires par les cellules épithéliales respiratoires, les cellules inflammatoires et immunitaires, et interviennent à différentes étapes de la cascade allergique. Ainsi, les HAP dérivés des particules diesel augmentent la production d’IgE par les lymphocytes B humains cultivés en présence d’IL-4 et d’anticorps anti-CD40 [11]. Les particules diesel augmentent l’adhérence des polynucléaires éosinophiles humains aux cellules épithéliales nasales, et induisent leur dégranulation [2]. Il existe un effet synergique des particules diesel avec l’allergène pour stimuler la production d’IL-8, de RANTES (regulated on activation, normal T expressed and secreted) et de TNFα par les cellules mononucléées sanguines périphériques de sujets allergiques [12]. L’exposition de cellules épithéliales bronchiques provenant de sujets asthmatiques à de faibles doses de particules diesel augmentent la production d’IL-8, de GM-CSF (granulocyte-macrophage colony stimulating factor) et de RANTES, tandis que de fortes doses sont nécessaires pour induire une augmentation significative de la production de ces médiateurs par des cellules de sujets non asthmatiques [2].
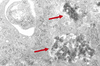 |
Figure 1. Macrophage humain dérivé des monocytes sanguins après 24 heures d’exposition en culture à des particules diesel [10]. L’analyse en microscopie électronique montre la localisation intracytoplasmique des particules diesel (➞) (agrandissement × 12 000). |
Les études réalisées in vivo chez l’homme confirment ces effets pro-inflammatoires des particules diesel (Tableau I). L’exposition contrôlée de sujets sains aux particules diesel induit une réponse inflammatoire, notamment par l’infiltration des muqueuses respiratoires par des polynucléaires neutrophiles, des lymphocytes B et des macrophages. Ces effets pro-inflammatoires pourraient amplifier l’inflammation observée dans l’asthme allergique [1, 2, 13, 14]. En effet, les études réalisées en chambre d’exposition montrent que les particules diesel augmentent l’hyperréactivité bronchique des sujets asthmatiques [2]. De nombreux arguments fondés sur les résultats des études d’exposition chez l’homme suggèrent que les particules diesel favorisent les réponses de type allergique. Lors de tests de provocation nasale allergénique chez les sujets allergiques, les taux d’IgE spécifiques antiallergéniques sont 20 à 50 fois plus élevés chez les sujets exposés à l’allergène en présence de particules diesel que chez les sujets exposés à l’allergène seul. Alors que l’exposition aux particules diesel seules stimule la production de nombreux types de cytokines, l’exposition combinée particules diesel + allergènes induit l’expression d’un profil de cytokines de type TH2 et entraîne la diminution de l’expression de l’IFNγ, une cytokine de type TH1 [1, 2]. Les concentrations d’histamine nasale après test de provocation allergénique sont multipliées par 3 lorsque l’allergène est administré en présence de particules diesel [1, 2]. D’autres travaux ont montré l’implication des particules diesel dans l’induction de sensibilisation primaire à l’allergène (Figure 2) [1, 15]. Chez des sujets atopiques, l’exposition nasale répétée à un antigène connu pour son grand caractère immunogène, l’hémocyanine de mollusque KLH (keyhole limpet hemocyanin), induit la production locale d’IgG et d’IgA anti-KLH, mais pas d’IgE. En revanche, l’exposition préliminaire aux particules diesel avant chaque exposition nasale au KLH induit la production locale d’IgE anti-KLH chez 60 % des sujets. La réexposition secondaire de ces sujets « répondeurs » au KLH plusieurs mois après l’exposition primaire entraîne des symptômes de rhinite et la production d’IgE anti-KLH.
 |
Figure 2. Modifications de la réponse immunitaire aux allergènes en présence de polluants atmosphériques. Les polluants (particules diesel, par exemple) sont internalisés par les cellules épithéliales respiratoires et les macrophages. La production de dérivés oxygénés induite par les polluants participe à l’augmentation du stress oxydant au niveau de la muqueuse respiratoire et entraîne la production de cytokines pro-inflammatoires. Dans ces conditions, il semble exister une facilitation de l’étape de présentation antigénique, notamment grâce à la diminution de la production d’IL-10 et à l’augmentation de l’expression de molécules de costimulation (CD80, CD86) par les cellules présentatrices de l’antigène, ces cellules entraînant le développement d’une réponse de type TH2. Les particules diesel favorisent la commutation isotypique vers la production d’IgE par les lymphocytes B. Les IgE spécifiques de l’allergène produites par les plasmocytes « sensibilisent » les mastocytes/basophiles. Après exposition à l’allergène, les mastocytes dégranulent et produisent notamment de l’IL-4, qui agit par rétrocontrôle positif sur les lymphocytes B et T. Les particules diesel ont également un effet direct sur la dégranulation et la production de cytokines par les mastocytes/basophiles. L’augmentation du stress oxydant au niveau de la muqueuse respiratoire et la production de cytokines pro-inflammatoires favorisent l’inflammation et l’hyperréactivité bronchique (d’après [1]). |
Ozone
L’ozone retrouvé dans l’air ambiant provient de réactions photochimiques impliquant les rayons solaires ultraviolets et les mélanges de dioxyde d’azote (NO2) et d’hydrocarbures produits par les véhicules à moteurs et les industries. L’ozone est probablement la source principale d’oxydants de la pollution atmosphérique des villes au climat ensoleillé, comme celles du pourtour méditerranéen ou de la Californie, par exemple.
De nombreuses études réalisées in vitro ou chez l’animal ont évalué les effets inflammatoires de l’ozone sur les voies aériennes [16, 17]. Quant aux connaissances sur les effets néfastes de l’ozone sur les maladies respiratoires, elles proviennent pour la plupart d’études d’exposition réalisées chez le sujet sain ou asthmatique.
L’exposition à des concentrations d’ozone inférieures à 0,5 ppm, en l’absence d’exercice physique, n’entraîne pas d’effet sur la fonction respiratoire. En revanche, l’exposition à l’ozone lors d’un exercice musculaire diminue les performances ventilatoires (diminution de la capacité vitale forcée et du volume expiré maximal seconde) et augmente la résistance des voies aériennes et la dyspnée [18, 19]. Un des effets les plus importants de l’ozone chez les sujets asthmatiques est probablement l’aggravation de l’inflammation chronique des voies aériennes [20]. De nombreuses études suggèrent que les effets de l’ozone sur l’obstruction bronchique sont beaucoup plus marqués chez les patients asthmatiques que chez les sujets sains.
L’exposition à l’ozone augmente la réponse immédiate et retardée à l’allergène chez les sujets asthmatiques [21, 22]. L’ozone induit une augmentation de la production intracellulaire de dérivés oxygénés et de la perméabilité des cellules épithéliales, qui pourraient favoriser la pénétration des allergènes inhalés dans la muqueuse respiratoire et la production de cytokines inflammatoires. Le TNFα joue un rôle central dans l’initiation de l’inflammation des voies aériennes et l’induction de l’hyperréactivité bronchique. Le niveau de stress oxydant produit par l’ozone induit une inflammation relayée par le TNFα qui dépend à la fois de l’efficacité des systèmes anti-oxydants et des concentrations d’ozone. Récemment, il a été montré qu’un variant du promoteur du gène du TNFα (TNF-308GG) pouvait diminuer le risque de survenue d’asthme chez l’enfant, mais que cette protection dépendait de la concentration en ozone dans l’environnement [23] : les enfants vivant dans les zones à forte exposition à l’ozone ne seraient pas protégés par le génotype GG. De plus, l’expression de variants non fonctionnels de gènes codants pour des enzymes anti-oxydantes (gluthation S-transférases M1 et P1) diminuerait la protection apportée par le génotype GG dans les zones à fortes concentrations d’ozone.
Dioxyde de soufre et dioxyde d’azote
L’exposition au dioxyde de soufre induit une bronchoconstriction rapide chez les sujets sains et asthmatiques. Récemment, une association entre un polymorphisme d’un promoteur du gène du TNFα et la susceptibilité de sujets asthmatiques au dioxyde de soufre inhalé (baisse de plus de 12 % du volume expiré maximal seconde) a été décrite [24].
Le dioxyde d’azote, produit par les véhicules à moteur et l’industrie, est un précurseur de la pollution photo-oxydante, dont les effets sur la santé sont principalement dus à la formation d’ozone. Une étude épidémiologique suggère que l’exposition au dioxyde d’azote est associée à une augmentation de la prévalence de l’asthme et de la rhinite allergique [25]. Chez la souris, l’exposition au dioxyde d’azote augmente la réponse inflammatoire de type allergique et l’hyperréactivité bronchique [26]. L’exposition au dioxyde d’azote pourrait favoriser la sensibilisation aux allergènes inhalés. Une exposition à 0,4 ppm de dioxyde d’azote pendant 4 heures augmenterait la réponse immédiate et retardée à l’allergène chez des sujets asthmatiques [27].
Mécanismes d’action et perspectives thérapeutiques
L’homme n’est pas allergique à la pollution, stricto sensu, dans la mesure où il ne développe pas de réponse immunitaire spécifique. Dans ces conditions, l’objectif des travaux expérimentaux est de mieux comprendre les interactions entre polluants atmosphériques et muqueuses respiratoires. Ces interactions peuvent en effet modifier la réponse immunitaire acquise et favoriser le développement de pathologies de type allergique, notamment. On distingue habituellement l’effet irritant, lié à l’exposition aiguë aux polluants, de leur effet à plus ou moins long terme sur le système immunitaire. Ces effets ne sont pas forcément indépendants, dans la mesure où les mécanismes d’irritation ont un retentissement plus important chez les sujets présentant une maladie respiratoire sous-jacente, qu’elle soit allergique ou non.
L’asthme est une maladie inflammatoire chronique des bronches qui met en jeu des réponses lymphocytaires de type TH2 et implique de nombreux types de cellules. Alors que le rôle des cytokines pro-inflammatoires, des chimiokines et des médiateurs des polynucléaires éosinophiles dans les réponses inflammatoires observées dans l’asthme et la rhinite allergique a été largement exploré [28], le rôle clé des oxydants n’est reconnu que de manière plus récente [1]. L’effet du stress oxydant sur la genèse de l’inflammation semble jouer un rôle important dans les effets délétères des polluants sur la santé. Li et ses collaborateurs [29] ont récemment décrit un modèle dans lequel des doses croissantes de particules de la pollution atmosphérique, puissants inducteurs du stress oxydant, entraînent une réaction de protection, puis une agression cellulaire. Le stress oxydant induit des voies de signalisation sensibles au statut redox de la cellule (MAP kinase [mitogen-activated protein kinase] et cascade du NF-κB [nuclear factor κB] qui activent l’expression de cytokines pro-inflammatoires, de chimiokines et de récepteurs de molécules d’adhérence. Cette activation intervient grâce à la mise en jeu de séquences stimulatrices se situant dans la région promotrice des gènes (genetic response elements). Selon ce modèle, à un niveau faible de stress oxydant, les particules de la pollution atmosphérique induisent des réponses protectrices par l’activation d’un promoteur de gène anti-oxydant (ARE, antioxidant response element) : cette activation nécessite l’expression et la translocation nucléaire du facteur de transcription Nrf-2 (NFE2p45-related factor-2), où il interagit avec l’ARE pour induire l’expression d’anti-oxydants (heme-oxygenase) et d’enzymes de phase II du métabolisme des xénobiotiques (glutathion-S-transférases, NADPH quinone oxydoréductase), qui possèdent des propriétés cytoprotectrices, anti-oxydantes et détoxifiantes. Si cette barrière de protection est dépassée, une nouvelle escalade du stress oxydant conduit à l’activation de la voie MAP-kinase/NF-κB et à des effets pro-inflammatoires [30]. Au plus haut degré de stress oxydant, les perturbations de la fonction mitochondriale aboutissent à l’apoptose de la cellule ou à sa nécrose. Ce modèle de hiérarchisation du stress oxydant prédit qu’une défaillance du système de défense anti-oxydant pourrait favoriser l’inflammation des voies aériennes induite par les particules et accroître la susceptibilité d’un individu à développer une maladie asthmatique [1, 31, 32]. Cette hypothèse permettrait d’expliquer l’existence de groupes de sujets à haut risque d’asthme lors de l’exposition à des polluants atmosphériques, en comparaison de sujets présentant de bonnes défenses anti-oxydantes. Plusieurs études ont démontré que les individus présentant des variants non fonctionnels des gènes d’enzymes de phase II sont plus sensibles aux effets adjuvants « pro-allergiques » des particules diesel et de l’ozone [1, 33, 34], ce qui illustre l’importance des gènes d’enzymes de phase II dans la régulation des réponses inflammatoires aux polluants. Plus récemment, Wan et ses collaborateurs [35] ont montré que l’induction d’une enzyme de phase II, la NADPH quinone oxydoréductase (NQO1), bloquait la surproduction d’IgE synthétisées par des lymphocytes B exposés aux particules diesel : ces résultats pourraient permettre d’envisager de nouvelles stratégies thérapeutiques préventives grâce à la surexpression d’enzymes de phase II. Parallèlement, différents travaux ont montré que la production de cytokines pro-inflammatoires par des cellules épithéliales bronchiques humaines stimulées par les particules diesel pouvait être inhibée par un prétraitement anti-oxydant [1, 2].
D’autres mécanismes concernant le rôle des polluants dans l’induction des allergies respiratoires ne seront pas discutés ici : il s’agit notamment de l’irritation de la muqueuse respiratoire et l’altération de la clairance mucociliaire, qui peuvent faciliter la pénétration des allergènes inhalés dans les voies aériennes et leur présentation aux cellules du système immunitaire [27]. Les polluants atmosphériques pourraient également modifier les propriétés allergéniques des particules transportant les pneumallergènes.
Conclusions
Alors que les premières études épidémiologiques mettaient en évidence de simples associations, les données scientifiques récentes apportent des éléments de réponse sur le rôle des polluants atmosphériques et leur mécanismes d’action dans les maladies allergiques respiratoires. Les polluants particulaires et gazeux peuvent favoriser ou aggraver les mécanismes physiopathologiques à l’origine de l’asthme ou de la rhinite allergique. Les avancées les plus importantes ont été réalisées pour la pollution particulaire, liée principalement aux émissions des véhicules à moteur diesel. Parmi les mesures préventives découlant de la connaissance des liens entre pollution atmosphérique et maladies allergiques respiratoires, citons la limitation des activités extérieures des sujets présentant un asthme ou une rhinite allergique pendant les périodes de forte pollution, la limitation des émissions de polluants, notamment des véhicules à moteur diesel, et le développement des études permettant de mieux caractériser les sujets plus sensibles aux effets irritants ou adjuvants des polluants, mais aussi de mieux préciser l’éventuelle place des traitements anti-oxydants dans la prévention des pathologies allergiques respiratoires.
Article reçu le 19 septembre 2006, accepté le 9 octobre 2006.
Références
- Saxon A, Diaz-Sanchez D. Air pollution and allergy : you are what you breathe. Nat Immunol 2005; 6 : 223–6. [Google Scholar]
- Riedl M, Diaz-Sanchez D. Biology of diesel exhaust effects on respiratory function. J Allergy Clin Immunol 2005; 115 : 221–8. [Google Scholar]
- Brauer M, Hoek G, Van Vliet P, et al. Air pollution from traffic and the development of respiratory infections and asthmatic and allergic symptoms in children. Am J Respir Crit Care Med 2002; 166 : 1092–8. [Google Scholar]
- Wyler C, Braun-Fahrlander C, Kunzli N, et al. Exposure to motor vehicle traffic and allergic sensitization. Epidemiology 2000; 11 : 450–6. [Google Scholar]
- Janssen NAH, Brunekreef B, Van Vliet P, et al. The relationship between air pollution from heavy traffic and allergic sensitization, bronchial hyperresponsiveness, and respiratory symptoms in Dutch schoolchildren. Environ Health Perspect 2003; 111 : 1512–8. [Google Scholar]
- Matsumoto A, Hiramatsu K, Li Y, et al. Repeated exposure to low-dose diesel exhaust after allergen challenge exaggerates asthmatic responses in mice. Clin Immunol 2006; 121 : 227–35. [Google Scholar]
- Finkelman FD, Yang M, Orekhova T, et al. Diesel exhaust particles suppress in vivo interferon γ production by inhibiting cytokine effects on NK and NKT cells. J Immunol 2004; 172 : 3808–13. [Google Scholar]
- Ohta K, Yamashita N, Tajima M, et al. Diesel exhaust particulate induces airway hyperresponsiveness in a murine model: essential role of GM-CSF. J Allergy Clin Immunol 1999; 104 : 1024–30. [Google Scholar]
- Kobayashi T. Exposure to diesel exhaust aggravates nasal allergic reaction in guinea pig. Am J Respir Crit Care Med 2000; 162 : 352–6. [Google Scholar]
- Bonay M, Chambellan A, Grandsaigne M, et al. Effects of diesel exhaust particles on the control of intracellular mycobacterial growth by human macrophages in vitro. FEMS Immunol Med Microbiol 2006; 46 : 419–25. [Google Scholar]
- Takenaka H, Zhang K, Diaz-Sanchez D, et al. Enhanced human IgE production results from exposure to the aromatic hydrocarbons from diesel exhaust: direct effects on B-cell IgE production. J Allergy Clin Immunol 1995; 95 : 103–15. [Google Scholar]
- Fahy O, Hammad H, Senechal S, et al. Synergistic effect of diesel organic extracts and allergen Der p 1 on the release of chemokines by peripheral blood mononuclear cells from allergic subjects: involvement of the MAP kinase pathway. Am J Respir Cell Mol Biol 2000; 23 : 247–54. [Google Scholar]
- Salvi SS, Nordenhall C, Blomberg A, et al. Acute exposure to diesel exhaust increases IL-8 and GRO-alpha production in healthy human airways. Am J Respir Crit Care Med 2000; 161 : 550–7. [Google Scholar]
- Stenfors N, Nordenhall C, Salvi SS, et al. Different inflammatory responses in asthmatic and healthy humans exposed to diesel. Eur Respir J 2004; 23 : 82–6. [Google Scholar]
- Diaz-Sanchez D, Garcia M P, Wang M, et al. Nasal challenge with diesel exhaust particles can induce sensitization to a neoallergen in the human mucosa. J Allergy Clin Immunol 1999; 104 : 1183–8. [Google Scholar]
- Kafoury RM, Pryor WA, Squadrito GL, et al. Induction of inflammatory mediators in human airway epithelial cells by lipid ozonation products. Am J Respir Crit Care Med 1999; 160 : 1934–42. [Google Scholar]
- Wagner JG, Van Dyken SJ, Wierenga JR, et al. Ozone exposure enhances endotoxin-induced mucous cell metaplasia in rat pulmonary airways. Toxicol Sci 2003; 74 : 437–46. [Google Scholar]
- McConnell R, Berhane K, Gilliland F, et al. Asthma in exercising children exposed to ozone: a cohort study. Lancet 2002; 359 : 386–91. [Google Scholar]
- Cartellejos M, Gold DR, Damokosh AI, et al. Acute effects of ozone on the pulmonary function of exercising school children from Mexico City. Am J Respir Crit Care Med 1995; 152 : 1501–7. [Google Scholar]
- Vagaggini B, Taccola M, Cianchetti S, et al. Ozone exposure increases eosinophilic airway response induced by previous allergen challenge. Am J Respir Crit Care Med 2002; 16 : 1073–7. [Google Scholar]
- Ball BA, Folinsbee LJ, Peden DB, et al. Allergen bronchoprovocation of patients with mild allergic asthma after ozone exposure. J Allergy Clin Immunol 1996; 98 : 563–72. [Google Scholar]
- Jorres R, Nowak D, Magnussen H. Effect of ozone exposure on allergen responsiveness in subjects with asthma or rhinitis. Am J Respir Crit Care Med 1996; 153 : 56–64. [Google Scholar]
- Li YF, Gauderman WJ, Avol E, et al. Associations of tumor necrosis factor G-308A with childhood asthma and wheezing. Am J Respir Crit Care Med 2006; 173 : 970–6. [Google Scholar]
- Winterton DL, Kaufman J, Keener CV, et al. Genetic polymorphisms as biomarkers of sensitivity to inhaled sulphur dioxide in subjects with asthma. Ann Allergy Asthma Immunol 2001; 86 : 232–8. [Google Scholar]
- De Marco R. The impact of climate and traffic related NO2 on the prevalence of asthma and allergic rhinitis in Italy. Clin Exp Allergy 2002; 32 : 1405–12. [Google Scholar]
- Poynter ME, Persinger RL, Irvin CG, et al. Nitrogen dioxide enhances allergic airway inflammation and hyperresponsiveness in the mouse. Am J Physiol Lung Cell Mol Physiol 2006; 290 : L144–52. [Google Scholar]
- D’Amato G, Liccardi G, D’Amato M, Holgate S. Environmental risk factors and allergic bronchial asthma. Clin Exp Allergy 2005; 35 : 1113–24. [Google Scholar]
- Bonay M, Neukirch C, Grandsaigne M, et al. Changes in airway inflammation following nasal allergic challenge in patients with seasonal rhinitis. Allergy 2006; 61 : 111–8. [Google Scholar]
- Li N, Hao M, Phalen RF, et al. Particulate air pollutants and asthma. A paradigm for the role of oxidative stress in PM-induced adverse health effects. Clin Immunol 2003; 109 : 250–65. [Google Scholar]
- Xiao GG, Wang M, Li N, Nel AE. Use of proteomics to demonstrate a hierarchical oxidative stress response to diesel exhaust particles in a macrophage cell line. J Biol Chem 2003; 278 : 50781–90. [Google Scholar]
- Bucchieri F, Puddicombe SM, Lordan JL, et al. Asthmatic bronchial epithelium is more susceptible to oxidant-induced apoptosis. Am J Respir Cell Mol Biol 2002; 27 : 179–85. [Google Scholar]
- Gilmour MI, Jaakkola MS, London SJ, et al. How exposure to environmental tobacco smoke, outdoor air pollutants, and increased pollen burdens influences the incidence of asthma. Environ Health Perspect 2006; 114 : 627–33. [Google Scholar]
- Gilliland FD, Li YF, Saxon A, Diaz-Sanchez D. Effect of glutathione-S-transferase M1 and P1 genotypes on xenobiotic enhancement of allergic responses: randomised, placebo- controlled crossover study. Lancet 2004; 363 : 119–25. [Google Scholar]
- Romieu I, Sienra-Monge JJ, Ramirez-Aguilar M, et al. Genetic polymorphism of GSTM1 and antioxidant supplementation influence lung function in relation to ozone exposure in asthmatic children in Mexico City. Thorax 2004; 59 : 8–10. [Google Scholar]
- Wan J, Diaz-Sanchez D. Phase II enzymes induction blocks the enhanced IgE production in B cells by diesel exhaust particles. J Immunol 2006; 177 : 3477–83. [Google Scholar]
Liste des tableaux
Effets adjuvants des particules diesel sur la réponse inflammatoire allergique. GM-CSF : granulocyte macrophage colony stimulating factor ; TH2 : T helper de type 2 ; RANTES : regulation upon activation normal T cell expressed and secreted ; MCP-3 : macrophage chemoattractant protein-3 ; MIP-1α : macrophage inflammatory protein-1α ; KLH : keyhole limpet hemocyanin.
Liste des figures
|
Figure 1. Macrophage humain dérivé des monocytes sanguins après 24 heures d’exposition en culture à des particules diesel [10]. L’analyse en microscopie électronique montre la localisation intracytoplasmique des particules diesel (➞) (agrandissement × 12 000). |
| Dans le texte | |
|
Figure 2. Modifications de la réponse immunitaire aux allergènes en présence de polluants atmosphériques. Les polluants (particules diesel, par exemple) sont internalisés par les cellules épithéliales respiratoires et les macrophages. La production de dérivés oxygénés induite par les polluants participe à l’augmentation du stress oxydant au niveau de la muqueuse respiratoire et entraîne la production de cytokines pro-inflammatoires. Dans ces conditions, il semble exister une facilitation de l’étape de présentation antigénique, notamment grâce à la diminution de la production d’IL-10 et à l’augmentation de l’expression de molécules de costimulation (CD80, CD86) par les cellules présentatrices de l’antigène, ces cellules entraînant le développement d’une réponse de type TH2. Les particules diesel favorisent la commutation isotypique vers la production d’IgE par les lymphocytes B. Les IgE spécifiques de l’allergène produites par les plasmocytes « sensibilisent » les mastocytes/basophiles. Après exposition à l’allergène, les mastocytes dégranulent et produisent notamment de l’IL-4, qui agit par rétrocontrôle positif sur les lymphocytes B et T. Les particules diesel ont également un effet direct sur la dégranulation et la production de cytokines par les mastocytes/basophiles. L’augmentation du stress oxydant au niveau de la muqueuse respiratoire et la production de cytokines pro-inflammatoires favorisent l’inflammation et l’hyperréactivité bronchique (d’après [1]). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.