")
")
| Issue |
Med Sci (Paris)
Volume 22, Number 2, Février 2006
|
|
|---|---|---|
| Page(s) | 178 - 182 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/2006222178 | |
| Published online | 15 February 2006 | |
Hypervariabilité génotypique des mélanomas : Un défi thérapeutique
Genotypic variability of melanoma: a therapeutic challenge
1
Service de Dermatologie, Hôpital de l’Hôtel-Dieu, 1, place de l’Hôpital, 69288 Lyon Cedex 02, France
2
Services de Dermatologie et de Génétique clinique, Hôpital de l’Hôtel-Dieu, 1, place de l’Hôpital, 69288 Lyon Cedex 02, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
On estime à environ 5 000 le nombre annuel de nouveaux cas de mélanome en France. La proportion des formes familiales est évaluée, selon les séries, entre 5 % et 10 %. Les aspects cliniques, histologiques et biologiques de progression du mélanome sont actuellement mieux connus ; les développements technologiques récents ont, quant à eux, permis d’aborder les mécanismes moléculaires de la progression tumorale et de mettre en lumière des gènes impliqués dans cette évolution. Toutefois, des difficultés importantes persistent : il existe une grande variabilité dans l’expression de ces gènes, non seulement d’un patient à l’autre, mais aussi selon les stades de la maladie, locaux ou métastatiques ; par ailleurs, certaines des mutations retrouvées au cours du mélanome peuvent également être présentes dans des lésions bénignes (nævus). La variabilité génotypique associée au mélanome rend le ciblage thérapeutique complexe, et constitue actuellement un défi majeur en termes de traitement. Cet article, volontairement non exhaustif, insistera surtout sur les anomalies génomiques les plus étudiées, desquelles semblent naître des perspectives thérapeutiques intéressantes.
Abstract
Cutaneous melanoma remains a management challenge. Melanoma is the leading cause of death from skin tumors worldwide. Melanoma progression is well defined in its clinical, histopathological and biological aspects, but the molecular mechanism involved and the genetic markers associated to metastatic dissemination are only beginning to be defined. The recent development of high-throughput technologies aimed at global molecular profiling of cancer is switching on the spotlight at previously unknown candidate genes involved in melanoma. Among those genes, BRAF is one of the most supposed to be of interest and targeted therapies are ongoing in clinical trials. In familial melanoma, germline mutations in two genes, CDKN2A and CDK4, that play a pivotal role in controlling cell cycle and division. It is hope that this better understanding of the biologic features of melanoma and the mechanisms underlying tumor-induced immunosuppression will lead to efficaceous targeted therapy.
© 2006 médecine/sciences - Inserm / SRMS
Le cancer représente l’une des principales causes de mortalité dans les pays industrialisés. La cancérogenèse est due au dérèglement de l’expression des gènes impliqués dans la prolifération, la survie ou la mort cellulaire, une dérégulation secondaire à la survenue de mutations de ces gènes. La plupart des cancers sont sporadiques, c’est-à-dire que les mutations sont apparues de façon somatique et n’existent qu’au niveau de la tumeur. Cependant, environ 5 % des cancers sont secondaires à une mutation initiale présente de façon constitutive chez l’individu : on parle alors de prédisposition génétique au cancer. Les gènes impliqués dans ces cancers familiaux peuvent également être à l’origine de cancers sporadiques, la mutation n’étant alors pas germinale, mais apparue au niveau de certains tissus, c’est-à-dire somatique. L’étude des fonctions des protéines, produits de ces gènes, ainsi que de leurs ligands permet d’améliorer les connaissances sur les mécanismes de prolifération cellulaire en général, et de la tumorogenèse en particulier. La découverte de mutations familiales autorise l’identification des individus à risque, permettant ainsi de mettre en place une stratégie thérapeutique ou préventive éventuelle, mais aussi de rassurer les patients indemnes de mutation.
Le mélanome est devenu un enjeu de santé publique majeur dans de nombreux pays : depuis le milieu des années 60, son incidence a en effet augmenté de 3 % à 8 % par an selon les pays. Malgré cette incidence accrue et une mortalité également en hausse, le taux de survie augmente : en 1960, 60 % des patients atteints de mélanome décédaient de leur maladie, contre 11 % actuellement. Cette amélioration est essentiellement due au progrès réalisés en termes de diagnostic précoce [1].
Mutations somatiques et mélanome
Différents types d’anomalies chromosomiques ou de niveaux d’expression génique ont été mis en évidence sur des échantillons de tumeur primitive de mélanome, notamment par hybridation génomique comparative (CGH) ou évaluation comparative du nombre de copies d’ADN par micro-arrays. Les anomalies les plus fréquemment retrouvées sont les délétions des chromosomes 1, 6, 9 et 10, et des translocations réciproques ou complexes entre les chromosomes 6, 7 et 8, dans plus de 95 % des cas [1–3]. Des corrélations génotype-phénotype existent, reliant le type anatomoclinique du mélanome (acral, superficiel, nodulaire, lentigineux) aux remaniements chromosomiques qui leur sont associés [4].
Le mélanome acral, qui intéresse essentiellement les zones non photo-exposées, est fréquemment porteur d’un nombre supérieur de remaniements en gain, c’est-à-dire de trisomies, lorsqu’il est comparé au mélanome d’extension superficielle [5]. Les mélanomes survenant en zones photo-exposées, quant à eux, présentent le plus souvent des aberrations sur les régions chromosomiques 13q et 17p [6]. De leur côté, les mélanomes atteignant les muqueuses sont surtout porteurs de translocations concernant les gènes régions 1q, 6p et 8q [7]. Les régions chromosomiques amplifiées contiennent le plus souvent des oncogènes connus (HRAS, CDK4, BRAF, cyclin D1) [8], mais également des gènes non identifiés [9]. Plusieurs études concordantes ont donc démontré que la nature des aberrations chromosomiques et le niveau d’expression de ces oncogènes sont corrélés au type anatomoclinique de mélanome et à sa survenue en zone photo-exposée ou non [4].
La séquence des modifications caryotypiques survenant au cours de l’évolution du mélanome est peu connue. Il semble toutefois que deux phénomènes initiaux, délétion du chromosome 3 ou translocation impliquant le chromosome 6p, sont essentiels avant que surviennent les autres altérations concernant les chromosomes 1, 8, 9, 10, 11 et 15 [10].
Le nombre d’aberrations chromosomiques, quant à lui, augmente avec l’évolution métastatique, traduisant une grande instabilité génétique. La comparaison génomique entre tumeur primitive et métastases révèle un grand nombre d’altérations accompagnant la progression de la maladie [11].
Gène BRAF
Une forte association entre des mutations du gène BRAF et le mélanome a été rapportée en 2002 par Davies et ses collaborateurs [12]. Il existe chez l’homme 3 gènes RAF (A-RAF, B-RAF et C-RAF) et 3 gènes RAS (H-RAS, K-RAS et N-RAS). Le gène BRAF code pour une sérine/thréonine kinase intervenant dans la voie de signalisation des MAP (mitogen-activated protein)-kinases (Figure 1). L’activation de cette voie conduit à des événements cytoplasmiques et transcriptionnels communs au développement de multiples cancers.
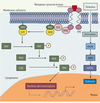 |
Figure 1. Voie de signalisation des MAP-kinases, avec les voies RAS-RAF-MEK-ERK. MAPK : mitogen-activated protein kinases ; MKK(K) : mitogen-activated protein kinase (kinase) ; RTK : receptor tyrosine kinase ; SH2 : sequence homology 2. |
Les gènes N-RAS et BRAF, impliqués dans la voie de signalisation des MAP-kinases, sont fréquemment mutés aux cours des stades locaux et métastatiques du mélanome. Les mutations de N-RAS sont présentes dans 5 % à 20 % des mélanomes. Quant aux mutations du gène BRAF, la plus commune est la mutation substitutive V600E (anciennement V599E), présente dans près de 66 % des cas de mélanome. Les mutations du gène BRAF semblent constituer un élément précoce dans l’évolution du mélanome, la progression métastatique de la maladie [13] et la prolifération vasculaire associée au développement tumoral [14] ; elles sont détectables dès la phase de croissance horizontale de ce cancer. Le transfert in vitro du gène BRAF muté conduit à des modifications du phénotype des mélanocytes, avec augmentation de leur prolifération et formation de tumeurs [15]. Des mutations du gène BRAF sont cependant également présentes au sein de nævus bénins. Les mutations germinales de ce gène semblent rares [16].
Associées à un mélanome, les mutations du gène BRAF sont plus fréquentes chez les sujets jeunes, lorsque le mélanome survient sur des zones d’exposition au soleil intense et intermittente (dos, jambes) [17]. Ces mutations sont rares, en revanche, sur les zones exposées de manière chronique aux radiations solaires (visage, extrémités des membres) [18], et absentes au cours des mélanomes oculaire [19] et desmoplastique [20].
In vitro, la suppression des produits d’expression du gène BRAF par interférence par l’ARN limite la prolifération cellulaire [21]. Les mutations du gène BRAF représentant une cible thérapeutique privilégiée, des agents interférant avec le gène BRAF sont en cours de développement clinique dans des essais thérapeutiques de phase III [22].
Gène MC1R
Le MC1-R, récepteur de l’α-MSH (mélanocortine) présent sur les mélanocytes, permet de stimuler la production de mélanines foncées (eumélanines) (Figure 2), qui sont photoprotectrices. Les mutations du gène MC1R entraînent une diminution de la production des eumélanines au profit de celle des phéomélanines, peu photoprotectrices [1], facilitant en cela la survenue de dommages photo-induits. Il existe un grand polymorphisme du gène MC1R dans la population caucasiennne : environ 30 variants ont été rapportés, parmi lesquel 9 sont associés à des pertes fonctionnelles [23, 24]. Des variants de MC1R ont été retrouvés en France chez 68 % des patients atteints de mélanome, contre 31 % dans un groupe contrôle. Les variants les plus significativement corrélés au risque de mélanome sont Val60Leu, Arg151Cys et Arg160Trp [25].
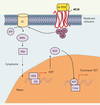 |
Figure 2. Régulation des gènes de la pigmentation de la peau par α-MSH /MC1R.Les effets de l’α-MSH sur la mélanogenèse passent par l’activation de la voie de l’AMPc. L’α-MSH se fixe sur son récepteur MC1R. Les protéines G transmettent le signal à l’AC, qui catalyse l’ATP en AMPc. L’augmentation de la concentration intracellulaire en AMPc provoque alors l’activation de la PKA, alors transloquée vers le noyau où elle phosphoryle le CREB. La présence d’un domaine CRE dans le promoteur de MITF explique son rôle central dans la régulation de l’expression des gènes de la pigmentation TRP1 et TRP2. α-MSH : α-melanocyte-stimulating hormone (mélanocortine) ; MC1R : melanocortin-1 receptor ; GPCR : G-protein-coupled receptor ; AC : adenylyl cyclase ; PKA : protein-kinase A ; CRE(B) : cAMP-responsive-element (binding protein) ; MITF : microphtalmia-associated transcription factor ; DCT : dopachrome tautomerase (ou TRP2) ; TRP1 : tyrosinase-related protein 1. |
Gène TP53
Le gène suppresseur de tumeur TP53, situé sur le bras court du chromosome 17, code pour une protéine nucléaire de 53 kDa (p53), un facteur de transcription intervenant à l’état normal dans le contrôle négatif du cycle cellulaire, la réparation et la division cellulaires, le contrôle de la stabilité génétique et l’apoptose. Quand le gène TP53 est muté, et la protéine inactive, les promoteurs de la croissance cellulaire s’expriment et déclenchent un développement anarchique de la cellule et, à terme, une tumeur cancéreuse.
Le rôle de p53 dans le mélanome n’est que partiellement compris. La protéine p53 est indétectable dans de nombreux tissus, y compris la peau, du fait de sa courte durée de vie. Cependant, l’étude en immuno-histochimie de tumeurs exprimant des variants mutés de p53 montre que cette protéine est stable dans le temps, et détectable. Il ne semble en revanche pas y avoir d’expression de p53 dans les nævus. Le niveau d’expression de la protéine p53, qui croît avec l’épaisseur de la lésion, est aussi plus élevé dans les métastases. Lorsque le mélanome survient sur un nævus, seules les cellules malignes expriment la protéine p53 [26]. Lors de l’analyse de la tumeur primitive, des pertes d’hétérozygotie (LOH) de TP53 ont été retrouvées dans environ 20 % des cas [27].
Gène PTEN
Le gène suppresseur de tumeur PTEN (phosphatase and tensin homolog deleted on chromosome TEN) est localisé sur le bras long du chromosome 10, en 10q23. Les mutations somatiques de ce gène ont pour conséquences la survenue de tumeurs cérébrales, de glioblastomes et de cancers de l’endomètre. Les mutations du gène PTEN sont fréquemment retrouvées dans les lignées cellulaires in vitro de mélanome. Une étude récente a retrouvé, sur des lésions primitives de mélanomes, jusqu’à 43 % de mutations de PTEN, sans que la présence ou l’absence de ces mutations soit liée au pronostic [28]. Lorsque la mutation du gène PTEN est germinale, les patients développent préférentiellement des cancers du sein et de la thyroïde, mais peu de mélanomes [29].
Gènes de prédisposition et mélanomes familiaux
Les formes familiales de cancer ont souvent permis de mettre en évidence des anomalies génétiques qui gouvernent à la fois les formes familiales et les formes sporadiques de la maladie. Les formes familiales représentent 8 % à 12 % des mélanomes. La proportion exacte des mélanomes associée à des gènes connus est mal évaluée, mais serait inférieure à 2 % [30]. Deux gènes majeurs de susceptibilité ont été identifiés en association avec les mélanomes familiaux : le gène suppresseur de tumeur CDKN2A et l’oncogène CDK4.
Le gène CDKN2A a été localisé en 9p21 grâce à des délétions homozygotes retrouvées fréquemment portées par des lignées tumorales [31, 32]. Ce locus, désigné ensuite comme associé aux mélanomes familiaux [33], code pour deux protéines, p16INK4a et p14ARF. La protéine p16INK4a est un inhibiteur de la phosphorylation de la pRB (produit du gène suppresseur de tumeur RB-1, pour rétinoblastome-1) par le complexe cycline D1-cdk4, inhibition qui entraîne un arrêt du cycle cellulaire en G1. La protéine p14ARF intervient, quant à elle, en inhibant la dégradation de la protéine p53 [34]. Une mutation du gène CDKN2A est présente dans 25 % à 40 % des formes familiales de mélanome, tandis qu’elle n’est que de 0,2 % à 2 % dans les formes sporadiques [35].
D’autres gènes sont en cours d’études ; ainsi, un gène candidat est localisé sur le bras court du chromosome 1, en 1p22 [36].
Conclusions et perspectives
À la suite du succès remarquable de l’inhibiteur sélectif de tyrosine-kinase (imatinib) dans le traitement de la leucémie myéloïde chronique, la cancérologie est entrée dans l’ère des traitements « de ciblage moléculaire ». Les anomalies chromosomiques et moléculaires au sein des tumeurs sont multiples et souvent variables pour une même tumeur, notamment au cours de son évolution métastatique. Il n’a pas encore été dégagé pour le mélanome de cible moléculaire essentielle au développement des mélanocytes tumoraux, spécifique du contingent cellulaire malin et persistant au cours de l’évolution métastatique. Il est probable que la solution émanera d’une thérapeutique ciblée, utilisée en monothérapie ou, plus probablement, en association, une thérapeutique visant non seulement les mélanocytes tumoraux, mais aussi l’environnement immédiat (en termes de vascularisation, de réponse immunitaire…) des cellules malignes, nécessaire à leur prolifération. La compréhension des anomalies génétiques et des mécanismes moléculaires impliqués dans le mélanome aux stades initiaux et disséminés progresse actuellement de manière spectaculaire. Les hypothèses de développements thérapeutiques sont aussi très nombreuses, même si le traitement du mélanome métastatique constitue encore un défi thérapeutique majeur pour les chercheurs et médecins impliqués dans la prise en charge de cette maladie.
Références
- Thompson JF, Scolyer RA, Kefford RF. Cutaneous melanoma. Lancet 2005; 365 : 687–701. [Google Scholar]
- Bastian BC. Understanding the progression of melanocytic neoplasia using genomic analysis : from fields to cancer. Oncogene 2003; 22 : 3081–6. [Google Scholar]
- Nelson MA, Radmacher MD, Simon R, et al. Chromosome abnormalities in malignant melanoma : clinical significance of nonrandom chromosome abnormalities in 206 cases. Cancer Genet Cytogenet 2000; 122 : 101–9. [Google Scholar]
- Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med 2005; 353 : 2135–47. [Google Scholar]
- Bastian BC, Kashani-Sabet M, Hamm H, et al. Gene amplifications characterize acral melanoma and permit the detection of occult tumor cells in the surrounding skin. Cancer Res 2000; 60 : 1968–73. [Google Scholar]
- Bastian BC, Olshen AB, LeBoit PE, Pinkel D. Classifying melanocytic tumors based on DNA copy number changes. Am J Pathol 2003; 163 : 1765–70. [Google Scholar]
- Van Dijk M, Sprenger S, Rombout P, et al. Distinct chromosomal aberrations in sinonasal mucosal melanoma as detected by comparative genomic hybridization. Genes Chromosomes Cancer 2003; 36 : 151–8. [Google Scholar]
- Chudnovsky Y, Adams AE, Robbins PB, et al. Use of human tissue to assess the oncogenic activity of melanoma-associated mutations. Nat Genet 2005; 37 : 745–9. [Google Scholar]
- Sauter ER, Yeo UC, von Stemm A, et al. Cyclin D1 is a candidate oncogene in cutaneous melanoma. Cancer Res 2002; 62 : 3200–6. [Google Scholar]
- Hoglund M, Gisselsson D, Hansen GB, et al. Dissecting karyotypic patterns in malignant melanomas : temporal clustering of losses and gains in melanoma karyotypic evolution. Int J Cancer 2004; 108 : 57–65. [Google Scholar]
- Balazs M, Adam Z, Treszl A, et al. Chromosomal imbalances in primary and metastatic melanomas revealed by comparative genomic hybridization. Cytometry 2001; 46 : 222–32. [Google Scholar]
- Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002; 417 : 949–54. [Google Scholar]
- Shinozaki M, Fujimoto A, Morton DL, Hoon DS. Incidence of BRAF oncogene mutation and clinical relevance for primary cutaneous melanomas. Clin Cancer Res 2004; 10 : 1753–7. [Google Scholar]
- Sharma A, Trivedi NR, Zimmerman MA, et al. Mutant V599EB-Raf regulates growth and vascular development of malignant melanoma tumors. Cancer Res 2005; 65 : 2412–21. [Google Scholar]
- Wellbrock C, Ogilvie L, Hedley D, et al. V599EB-RAF is an oncogene in melanocytes. Cancer Res 2004; 64 : 2338–42. [Google Scholar]
- Laud K, Kannengiesser C, Avril MF, et al. BRAF as a melanoma susceptibility candidate gene ? Cancer Res 2003; 63 : 3061–5. [Google Scholar]
- Kumar R, Angelini S, Czene K, et al. BRAF mutations in metastatic melanoma : a possible association with clinical outcome. Clin Cancer Res 2003; 9 : 3362–8. [Google Scholar]
- Maldonado JL, Fridlyand J, Patel H, et al. Determinants of BRAF mutations in primary melanomas. J Natl Cancer Inst 2003; 95 : 1878–90. [Google Scholar]
- Davison JM, Rosenbaum E, Barrett TL, et al. Absence of V599E BRAF mutations in desmoplastic melanomas. Cancer 2005; 103 : 788–92. [Google Scholar]
- Hingorani SR, Jacobetz MA, Robertson GP, et al. Suppression of BRAF(V599E) in human melanoma abrogates transformation. Cancer Res 2003; 63 : 5198–202. [Google Scholar]
- Bollag G, Freeman S, Lyons JF, Post LE. Raf pathway inhibitors in oncology. Curr Opin Investig Drugs 2003; 4 : 1436–41. [Google Scholar]
- Cruz F, Rubin BP, Wilson D, et al. Absence of BRAF and NRAS mutations in uveal melanoma. Cancer Res 2003; 63 : 5761–6. [Google Scholar]
- Schaffer JV, Bolognia JL. The melanocortin-1 receptor : red hair and beyond. Arch Dermatol 2001; 137 : 1477–1485. [Google Scholar]
- Sturm RA, Teasdale RD, Box NF. Human pigmentation genes : identification, structure and consequences of polymorphic variation. Gene 2001; 277 : 49–62. [Google Scholar]
- Matichard E, Verpillat P, Meziani R, et al. Melanocortin 1 receptor (MC1R) gene variants may increase the risk of melanoma in France independently of clinical risk factors and UV exposure. J Med Genet 2004; 41 : e13. [Google Scholar]
- Radhi JM. Malignant melanoma arising from nevi, p53, p16, and Bcl-2 : expression in benign versus malignant components. J Cutan Med Surg 1999; 3 : 293–7. [Google Scholar]
- Soto JL, Cabrera CM, Serrano S, Lopez-Nevot MA. Mutation analysis of genes that control the G1/S cell cycle in melanoma : TP53, CDKN1A, CDKN2A, and CDKN2B. BMC Cancer 2005; 5 : 36. [Google Scholar]
- Mikhail M, Velazquez E, Shapiro R, et al. PTEN expression in melanoma : relationship with patient survival, Bcl-2 expression, and proliferation. Clin Cancer Res 2005; 11 : 5153–7. [Google Scholar]
- Liaw D, Marsh DJ, Li J, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet 1997; 16 : 64–7. [Google Scholar]
- Goldstein AM, Tucker MA. Genetic epidemiology of cutaneous melanoma : a global perspective. Arch Dermatol 2001; 137 : 1493–6. [Google Scholar]
- Kamb A, Gruis NA, Weaver-Feldhaus J, et al. A cell cycle regulator potentially involved in genesis of many tumor types. Science 1994; 264 : 436–40. [Google Scholar]
- Nobori T, Miura K, Wu DJ, et al. Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature 1994; 368 : 753–6. [Google Scholar]
- Hussussian CJ, Struewing JP, Goldstein AM, et al. Germline p16 mutations in familial melanoma. Nat Genet 1994; 8 : 15–21. [Google Scholar]
- Chin L. The genetics of malignant melanoma : lessons from mouse and man. Nat Rev Cancer 2003; 3 : 559–70. [Google Scholar]
- Tsao H, Zhang X, Kwitkiwski K, et al. Low prevalence of germline CDKN2A and CDK4 mutations in patients with early-onset melanoma. Arch Dermatol 2000; 136 : 1118–22. [Google Scholar]
- Gillanders E, Juo SH, Holland EA, et al. Localization of a novel melanoma susceptibility locus to 1p22. Am J Hum Genet 2003; 73 : 301–13. [Google Scholar]
Liste des figures
|
Figure 1. Voie de signalisation des MAP-kinases, avec les voies RAS-RAF-MEK-ERK. MAPK : mitogen-activated protein kinases ; MKK(K) : mitogen-activated protein kinase (kinase) ; RTK : receptor tyrosine kinase ; SH2 : sequence homology 2. |
| Dans le texte | |
|
Figure 2. Régulation des gènes de la pigmentation de la peau par α-MSH /MC1R.Les effets de l’α-MSH sur la mélanogenèse passent par l’activation de la voie de l’AMPc. L’α-MSH se fixe sur son récepteur MC1R. Les protéines G transmettent le signal à l’AC, qui catalyse l’ATP en AMPc. L’augmentation de la concentration intracellulaire en AMPc provoque alors l’activation de la PKA, alors transloquée vers le noyau où elle phosphoryle le CREB. La présence d’un domaine CRE dans le promoteur de MITF explique son rôle central dans la régulation de l’expression des gènes de la pigmentation TRP1 et TRP2. α-MSH : α-melanocyte-stimulating hormone (mélanocortine) ; MC1R : melanocortin-1 receptor ; GPCR : G-protein-coupled receptor ; AC : adenylyl cyclase ; PKA : protein-kinase A ; CRE(B) : cAMP-responsive-element (binding protein) ; MITF : microphtalmia-associated transcription factor ; DCT : dopachrome tautomerase (ou TRP2) ; TRP1 : tyrosinase-related protein 1. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.