")
")
| Issue |
Med Sci (Paris)
Volume 22, Number 1, Janvier 2006
|
|
|---|---|---|
| Page(s) | 29 - 32 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/200622129 | |
| Published online | 15 janvier 2006 | |
Immunité innée et sélection naturelle : Le paradigme de la région génomique DC-SIGN / L-SIGN
Distinct functions of DC-SIGN and its homologues L-SIGN and mSIGNR1 in pathogen recognition and immune regulation
CNRS FRE2849, Unité de prévention et thérapie moléculaires des maladies humaines, Institut Pasteur, 25, rue du Docteur Roux, 75015 Paris, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
L’histoire de l’homme, c’est aussi l’histoire des pathogènes qui l’infectent et de leur co-évolution. Les maladies infectieuses ont certainement constitué un facteur de pression de sélection majeur dans le processus d’adaptation de notre espèce aux différents environnements dont font partie les pathogènes. Outre la biologie cellulaire, la génétique classique et l’épidémiologie, la génétique des populations et la biologie évolutive offrent des approches et stratégies alternatives pour mieux comprendre cette étroite relation entre l’homme et les microbes, et permettent d’évaluer les conséquences d’une exposition différentielle de l’homme aux environnements pathogènes sur la variabilité du génome humain. L’adaptation de l’homme aux pathogènes a entraîné des processus biologiques sélectifs qui peuvent avoir laissé des traces sur la répartition de la variabilité génétique dans les populations humaines [1]. En effet, outre les effets de la démographie et l’histoire des populations, la sélection naturelle est une des forces les plus importantes ayant influencé la variabilité du génome [2]. La sélection naturelle peut influencer, par exemple, la fréquence d’un variant génétique qui fait que les individus porteurs de celui-ci sont mieux protégés face à une maladie comme le paludisme. Ce variant aura donc la tendance à augmenter en fréquence dans une population habitant dans une région où le paludisme est endémique. Ainsi, à la différence des effets démographiques qui agissent de la même manière sur la totalité du génome, la sélection naturelle agit de manière ciblée et variable sur certaines parties du génome, selon leur fonction spécifique et leur rôle dans l’adaptation de l’homme à l’environnement.
De nombreuses études ont cherché les empreintes génétiques de la sélection naturelle sur des gènes humains impliqués dans l’immunité ou dans les relations hôte/pathogène. Ainsi, la sélection naturelle semble avoir laissé des traces sur des gènes humains comme ceux codant pour le système CMH, la β-globine, G6PD et CCR5, entre autres [3–6]. Cependant, très peu d’études se sont concentrées sur la variabilité génétique de gènes humains impliqués dans la reconnaissance précoce des agents infectieux (ou de leurs produits métaboliques), et encore moins sur l’impact de la sélection naturelle sur le système de la réponse immunitaire innée. En effet, l’immunité innée constitue la première ligne de défense et d’interaction entre l’hôte humain et les pathogènes [7]. Les cellules effectrices de l’immunité innée, comme les macrophages ou les cellules dendritiques, ont pour fonction de détecter les pathogènes en reconnaissant des motifs moléculaires exprimés par ces derniers. Ces cellules effectrices arborent des récepteurs qui reconnaissent comme des « corps étrangers » les motifs moléculaires caractérisant les pathogènes et déclenchent une série de réactions qui aboutissent finalement à une réponse immunitaire plus spécifique, dite réponse adaptative. Parmi ces récepteurs cellulaires, les lectines de type C ainsi que les récepteurs Toll-like ont fait l’objet de nombreuses études au cours des dernières années dans le domaine de l’immunologie, approfondissant nos connaissances sur les interactions précoces entre l’hôte humain et les agents infectieux [8].
Les lectines de type C : DC-SIGN et L-SIGN
Les lectines sont des protéines, membranaires ou solubles, qui reconnaissent des motifs glycosylés. On les dit « de type C » lorsque cette reconnaissance requiert des ions calcium. Plus particulièrement, deux membres de la famille des lectines de type C, DC-SIGN (dendritic cell-specific ICAM-3 grabbing nonintegrin) et L-SIGN (liver/lymph node-specific ICAM-3 grabbing nonintegrin), se sont révélés très intéressants dans le cadre des interactions hôte-pathogène [9]. Ces deux gènes sont localisés sur le bras court du chromosome 19 et résultent d’une duplication (Figure 1). Ils codent des récepteurs des cellules effectrices de l’immunité innée et ils interagissent avec un grand nombre d’agents infectieux dont certains d’une importance capitale en santé publique. Ainsi, DC-SIGN reconnaît des bactéries comme Mycobacterium tuberculosis et Helicobacter pylori, des virus comme VIH-1, Ebola, le virus de l’hépatite C et de la dengue, ainsi que des parasites comme Leishmania pifanoi et Schistosoma mansoni [9]. De son côté, L-SIGN interagit avec des virus comme celui de l’hépatite C, HIV, Ebola et le coronavirus, ainsi qu’avec des parasites comme Schistosoma mansoni, entre autres [9]. Afin de déterminer les niveaux de variabilité génétique de DC-SIGN et L-SIGN au sein de notre espèce ainsi que d’identifier dans quelle mesure, et comment, la sélection naturelle a pu influencer leur évolution, nous avons obtenu la séquence complète de ces deux gènes dans un panel multi-ethnique d’individus provenant d’Afrique sub-saharienne, d’Europe et d’Asie de l’Est [10].
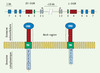 |
Figure 1. Organisation génomique et protéique de la région contenant DC-SIGN et L-SIGN. Les deux gènes sont localisés sur le bras court du chromosome 19 (19p13.2-3) et possèdent une structure génomique identique ainsi qu’une identité de séquence très élevée (73 %). Ils codent des récepteurs transmembranaires composés : (1) d’une partie extracellulaire qui contient le domaine de reconnaissance des hydrates de carbone caractérisant les pathogènes (CRD, carbohydrate recognition domain) et d’un domaine (neck-region) reliant le CRD à la région transmembranaire ; (2) d’un domaine transmembranaire (TM) ; et, enfin, (3) d’un domaine cytoplasmique caractérisé par la présence de plusieurs motifs, dont un motif tyrosine (Y), uniquement dans le cas de DC-SIGN, et des motifs tri-acidiques (EEE ou DEE) et di-leucine (LL). |
Différentes signatures moléculaires de la sélection naturelle…
Nous avons mis en évidence, par analyse de diversité nucléotidique, de coalescence et différents tests de neutralité, deux scénarios évolutifs opposés pour les deux lectines. Premièrement, DC-SIGN montre une diversité nucléotidique bien plus réduite que son homologue L-SIGN. Cette différence n’étant pas due à des taux mutationnels hétérogènes entre les deux gènes (l’estimation de ces taux de mutation donne des valeurs équivalentes), on peut envisager que des forces sélectives en sont responsables. En outre, DC-SIGN montre une réduction significative en mutations non-synonymes (mutations changeant l’acide aminé), ce qui suggère la présence d’une contrainte sélective sur ce gène qui empêcherait l’accumulation de changements au niveau protéique. À l’inverse, L-SIGN présente non seulement un excès de diversité de séquence (deux fois plus que la moyenne estimée pour l’ensemble du génome humain), mais aussi une diversité chez les Européens et les Asiatiques clairement plus élevée que dans les populations africaines. Cette dernière observation est en opposition avec la plupart des études qui montrent une plus grande diversité en Afrique qu’hors d’Afrique, soutenant une origine récente et africaine de notre espèce (modèle du remplacement rapide ou Out-of-Africa). Les données de variabilité observées dans L-SIGN, soutenues formellement par les différents tests de neutralité et distances génétiques entre populations, suggèrent ainsi que ce gène aurait été sous les effets de la sélection balancée (avantage à l’hétérozygote) en Europe et en Asie, ce qui expliquerait sa plus grande diversité observée dans ces populations.
…et la cause fonctionnelle de telles pressions de sélection ?
Les profils de variabilité génétique observés dans cette région génomique, suggérant des pressions de sélection différentielles pour les deux gènes, représentent la signature moléculaire de la sélection naturelle, plutôt que la cause fonctionnelle de celle-ci. Dans ce contexte, une partie de la protéine, dite neck-region, pourrait être la cible idéale de la sélection naturelle. En effet, le domaine extracellulaire de la protéine est constitué d’une partie ayant pour fonction la reconnaissance directe des pathogènes (carbohydrate recognition domain) et elle est elle-même reliée à la membrane cellulaire par la neck-region (Figure 1). Cette région, codée par l’exon 4 et présente dans les deux gènes, est normalement constituée de 7 répétitions qui codent chacune 23 acides aminés. Elle est directement impliquée dans la tétramérisation de la protéine et dans l’attachement aux pathogènes. Afin de mesurer le degré de polymorphisme de répétition de la neck-region dans DC-SIGN et L-SIGN, nous avons génotypé la totalité du panel multi-ethnique du CEPH (1064 individus provenant de 52 populations différentes). Nos données montrent que la neck-region de DC-SIGN possède un taux de polymorphisme extrêmement faible (hétérozygotie de 2 %), la grande majorité des individus (98 %) présentant 7 répétitions au niveau de cette région (Figure 2A). En revanche, la même région dans L-SIGN se caractérise par une variabilité générale très élevée (hétérozygotie 54 %) et, hors d’Afrique, les niveaux de polymorphisme de répétition deviennent plus importants que dans le continent africain (Figure 2B). Ainsi, l’ensemble de ces résultats suggère que la région répétée de l’exon 4, impliquée dans la capacité de ces lectines à reconnaître les pathogènes, pourrait être la cause fonctionnelle des patrons de sélection observés. En résumé, l’intégration des données de diversité nucléotidique et protéique, des tests de neutralité, des distances génétiques entre populations ainsi que du polymorphisme de longueur de la neck-region, montre que DC-SIGN a été sous contrainte sélective pendant l’évolution, ce qui soutient le rôle très important de cette lectine dans la reconnaissance des pathogènes et dans les premières phases de la réponse immunitaire. En revanche, ces pressions de sélection ont été très différentes pour son homologue L-SIGN, qui semble avoir été influencé plutôt par les effets de la sélection balancée, au moins en Europe et en Asie.
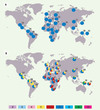 |
Figure 2. Distribution géographique des polymorphismes de répétition de la neck-region pour DC-SIGN (A) et L-SIGN (B). Les numéros (2 à 10) dans les rectangles en couleur correspondent à la longueur de la neck-region en nombre de répétitions. Les codes des populations sont : (1) Algériens ; (2) Mandenka ; (3) Yoruba ; (4) Pygmées Biaka ; (5) Bantous du Kenya ; (6) Pygmées Mbuti ; (7) San de Namibie ; (8) Bantous d’Afrique du Sud ; (9) Français et Basques ; (10) Italiens du Nord, du Centre et de Sardaigne (11) Orcadiens ; (12) Russes ; (13) Adygei ; (14) Druses, Palestiniens et Bédouins ; (15) Yakut ; (16) différentes populations Pakistanaises ; (17) différentes populations Chinoises ; (18) Japonais ; (19) Cambodgiens ; (20) Papous ; (21) Mélanésiens ; (22) Pima ; (23) Maya ; (24) Piapoco et Curripaco ; (25) Surui et (26) Karitiana. Pour tout détail sur la composition précise et l’origine de toutes les populations de cette collection, se reporter au site Internet du CEPH (http://www.cephb.fr/HGDP-CEPH-Panel/). |
Pour conclure
Cette étude illustre l’importance de la duplication de gènes de l’immunité comme stratégie développée par l’hôte pour mieux s’adapter et combattre les agents infectieux. En effet, le maintien, par conservation d’un duplicat génique, de la fonction basique de la protéine permet à son homologue de muter « librement » et d’acquérir de nouvelles fonctions éventuellement avantageuses. Cette stratégie serait d’autant plus attendue au sein des gènes de l’immunité car la co-évolution entre hôtes et pathogènes est très rapide et nécessite une très grande « flexibilité » évolutive. Plus généralement, l’étude de l’influence de la sélection naturelle sur la variabilité des gènes impliqués dans les interactions hôte-pathogène est une méthode alternative pour cibler les gènes qui ont été, et probablement sont encore, d’une grande importance pour la défense de l’homme contre les agents infectieux.
Remerciements
Je remercie Étienne Patin pour sa lecture critique du manuscrit.
Références
- Vallender EJ, Lahn BT. Positive selection on the human genome. Hum Mol Genet 2004; 13 (n° spécial) : R245–54. [Google Scholar]
- Bamshad M, Wooding SP. Signatures of natural selection in the human genome. Nat Rev Genet 2003; 4 : 99–111. [Google Scholar]
- Prugnolle F, Manica A, Charpentier M, et al. Pathogen-driven selection and worldwide HLA class I diversity. Curr Biol 2005; 15 : 1022–7. [Google Scholar]
- Flint J, Harding RM, Boyce AJ, Clegg JB. The population genetics of the haemoglobinopathies. Baillieres Clin Haematol 1998; 11 : 1–51. [Google Scholar]
- Sabeti PC, Reich DE, Higgins JM, et al. Detecting recent positive selection in the human genome from haplotype structure. Nature 2002; 419 : 832–7. [Google Scholar]
- Bamshad MJ, Mummidi S, Gonzalez E, et al. A strong signature of balancing selection in the 5’ cisregulatory region of CCR5. Proc Natl Acad Sci USA 2002; 99 : 10539–44. [Google Scholar]
- Janeway CA Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol 2002; 20 : 197–216. [Google Scholar]
- Gordon S. Pattern recognition receptors : doubling up for the innate immune response. Cell 2002; 111 : 927–30. [Google Scholar]
- Koppel EA, van Gisbergen KP, Geijtenbeek TB, van Kooyk Y. Distinct functions of DC-SIGN and its homologues L-SIGN (DC-SIGNR) and mSIGNR1 in pathogen recognition and immune regulation. Cell Microbiol 2005; 7 : 157–65. [Google Scholar]
- Barreiro LB, Patin E, Neyrolles O, Cann HM, Gicquel B, Quintana-Murci L. The heritage of pathogen pressures and ancient demography in the human innate-immunity CD209/CD209L region. Am J Hum Genet 2005; 77 : 869–86. [Google Scholar]
© 2006 médecine/sciences - Inserm / SRMS
Liste des figures
|
Figure 1. Organisation génomique et protéique de la région contenant DC-SIGN et L-SIGN. Les deux gènes sont localisés sur le bras court du chromosome 19 (19p13.2-3) et possèdent une structure génomique identique ainsi qu’une identité de séquence très élevée (73 %). Ils codent des récepteurs transmembranaires composés : (1) d’une partie extracellulaire qui contient le domaine de reconnaissance des hydrates de carbone caractérisant les pathogènes (CRD, carbohydrate recognition domain) et d’un domaine (neck-region) reliant le CRD à la région transmembranaire ; (2) d’un domaine transmembranaire (TM) ; et, enfin, (3) d’un domaine cytoplasmique caractérisé par la présence de plusieurs motifs, dont un motif tyrosine (Y), uniquement dans le cas de DC-SIGN, et des motifs tri-acidiques (EEE ou DEE) et di-leucine (LL). |
| Dans le texte | |
|
Figure 2. Distribution géographique des polymorphismes de répétition de la neck-region pour DC-SIGN (A) et L-SIGN (B). Les numéros (2 à 10) dans les rectangles en couleur correspondent à la longueur de la neck-region en nombre de répétitions. Les codes des populations sont : (1) Algériens ; (2) Mandenka ; (3) Yoruba ; (4) Pygmées Biaka ; (5) Bantous du Kenya ; (6) Pygmées Mbuti ; (7) San de Namibie ; (8) Bantous d’Afrique du Sud ; (9) Français et Basques ; (10) Italiens du Nord, du Centre et de Sardaigne (11) Orcadiens ; (12) Russes ; (13) Adygei ; (14) Druses, Palestiniens et Bédouins ; (15) Yakut ; (16) différentes populations Pakistanaises ; (17) différentes populations Chinoises ; (18) Japonais ; (19) Cambodgiens ; (20) Papous ; (21) Mélanésiens ; (22) Pima ; (23) Maya ; (24) Piapoco et Curripaco ; (25) Surui et (26) Karitiana. Pour tout détail sur la composition précise et l’origine de toutes les populations de cette collection, se reporter au site Internet du CEPH (http://www.cephb.fr/HGDP-CEPH-Panel/). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.