")
")
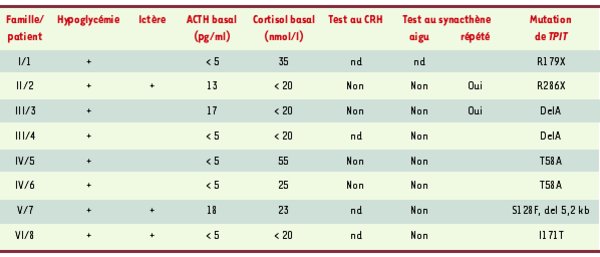
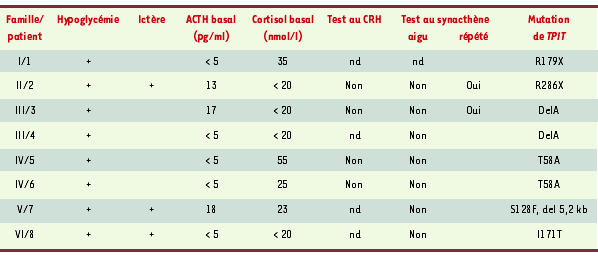
Tableau I.
Présentation clinique des huit patients atteints de déficit corticotrope isolé congénital et porteurs d’une mutation du gène TPIT. L’hypoglycémie néonatale était le plus souvent le premier symptôme ayant conduit au diagnostic de déficit en ACTH ; elle était le plus souvent sévère, quelquefois compliquée de convulsions. Trois patients ont présenté un ictère néonatal cholestatique prolongé. Tous les patients présentaient des concentrations plasmatiques effondrées d’ACTH (normale : 20-60 pg/ml) et de cortisol (normale : 250-600 nmol/l) ; les concentrations plasmatiques des autres hormones hypophysaires étaient normales dans tous les cas. La réponse au CRH de l’ACTH/cortisol (test au CRH) s’est révélée négative dans les quatre cas testés. L’injection aiguë de synacthène (test à l’ACTH) ne modifiait pas la concentration de cortisol plasmatique ; en revanche, les injections répétées de synacthène pendant 3 jours ont permis d’observer une réponse du cortisol dans deux cas, indiquant l’intégrité fonctionnelle des surrénales. nd : non déterminé.
{kind=link}
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.