")
")
| Issue |
Med Sci (Paris)
Volume 20, Number 3, Mars 2004
|
|
|---|---|---|
| Page(s) | 291 - 297 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/2004203291 | |
| Published online | 15 mars 2004 | |
L’agrégation plaquettaire et ses inhibiteurs dans les syndromes coronariens aigus
Platelet aggregation and antiplatelet agents in acute coronary syndromes
Institut de cardiologie, Hôpital de la Pitié-Salpêtrière, 47, boulevard de l’Hôpital, 75013 Paris, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Les antiagrégants plaquettaires sont des outils majeurs de la prise en charge des syndromes coronariens aigus. À la phase aiguë de l’infarctus avec élévation du segment ST, ils permettent de limiter l’effet prothrombogène des traitements de reperfusion tels que la thrombolyse (dissolution chimique du caillot obstruant l’artère coronaire) et l’angioplastie primaire (désobstruction mécanique de l’artère coronaire occluse). Dans le cadre des syndromes coronariens aigus sans élévation du segment ST, les antiagrégants plaquettaires permettent d’éviter l’évolution de la thrombose coronarienne partielle vers l’occlusion vasculaire complète, et donc vers l’infarctus aigu avec élévation du segment ST. Le bénéfice des antiagrégants plaquettaire dépend du profil de risque des patients ; il est démontré que l’association de plusieurs antiagrégants est d’autant plus efficace que les patients sont à plus haut risque. Les principaux enjeux de l’utilisation de cette classe thérapeutique sont les suivants : dépister les résistances, adapter les posologies et gérer l’arrêt temporaire de ces traitements chez des patients ayant une pathologie coronarienne stable.
Abstract
Antiplatelet agents are the cornerstone therapy of acute coronary syndromes. In the setting of ST elevation myocardial infarction, antiplatelet therapy prevent the prothrombotic effect of reperfusion therapy including thrombolysis and primary percutaneous coronary intervention. In non ST-elevation acute coronary syndromes, antiplatelet therapy prevent s complete coronary thrombotic occlusion and therefore the occurrence of ST elevation myocardial infarction. Antiplatelet agent benefit is related to the patient’s risk profile. It is well established that combined antiplatelet therapy is the most effective in high risk patients. Several important issues have to be faced including the identification of non responders, dose adjustment and the management of temporary interruption of antiplatelet agents in stable coronary artery disease patients.
© 2004 médecine/sciences - Inserm / SRMS
Physiopathologie des syndromes coronariens aigus
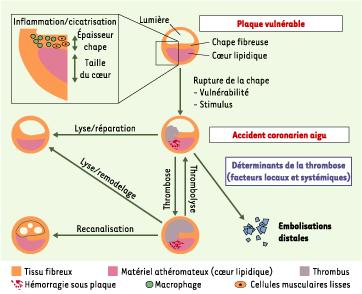
La gravité de l’athérosclérose coronarienne est surtout liée au risque permanent d’accident aigu (angor instable ou infarctus du myocarde) (Encadré 1) associé au déclenchement du processus thrombotique à la surface d’une plaque rompue. La rupture ou l’érosion de la plaque met en effet le sang circulant en contact avec les éléments thrombogènes du cœur lipidique, provoquant ainsi l’activation des plaquettes et de la coagulation, et la formation du thrombus (Encadré 2, Figure 1).
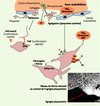 |
Figure 1. Formation de l’agrégat plaquettaire et du réseau de fibrine à la surface de la plaque rompue. Les plaquettes forment dans un premier temps un tapis au niveau de la plaque lésée. L’interaction de la glycoprotéine Iba (GP Ib) plaquettaire avec le facteur von Willebrand (FvW) est primordiale dans des conditions de contraintes de cisaillement élevées, par exemple en cas de sténose athéromateuse serrée dans un vaisseau de petit diamètre : elle permet en effet l’adhérence des plaquettes au sous-endothélium activé. L’activation plaquettaire survient dans un deuxième temps, et résulte soit de l’adhérence des plaquettes au sous-endothélium, soit de leur exposition à des agonistes circulants, notamment la thrombine et le collagène, premiers activateurs trouvés au niveau de la brèche vasculaire. La présence de facteur tissulaire (FT) dans des microparticules libérées par les cellules inflammatoires de la plaque entraîne l’activation de la coagulation. Le complexe Xa/Va (prothrombinase) permet la transformation de la prothrombine en thrombine (IIa) (le facteur VIIIa est un puissant cofacteur de la production de thrombine), à l’origine de la formation de fibrine à partir du fibrinogène. Le complexe GP IIb/IIIa est quant à lui responsable de l’agrégation plaquettaire par le fibrinogène, qui forme des ponts interplaquettaires (voir Figure 2). La sérotonine, le thromboxane A2 (TXA2) et l’ADP sont libérés par les plaquettes pour activer et recruter de nouvelles plaquettes. |
1. Classification nosologique des syndromes coronariens aigus (SCA) [2]
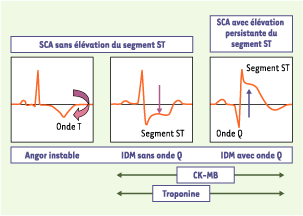
L’infarctus du myocarde (IDM), avec élévation du segment ST (flèche bleue, à droite), résulte d’une occlusion coronarienne complète. Il impose la mise en œuvre la plus rapide possible d’un traitement par thrombolyse ou par angioplastie transluminale, afin de désobstruer le vaisseau occlus. La thrombolyse, en libérant la thrombine piégée dans le thrombus en cours de dégradation, et l’angioplastie, par la fragmentation du caillot et de la plaque, favorisent la thrombogenèse. Les antiagrégants plaquettaires limitent l’effet prothrombogène de ces traitements. L’onde Q correspond à la cicatrice de l’infarctus du myocarde qui occupe toute l’épaisseur du muscle cardiaque.
Les SCA sans élévation du segment ST, plus fréquents, forment une entité hétérogène dont la prise en charge diagnostique est l’étape clé, afin d’individualiser les patients les plus à risque. On distingue l’infarctus sans onde Q (flèche mauve, au milieu), qui correspond à un infarctus sous-endocardique (avec élévation des marqueurs de nécrose myocardique CK-MB [créatinine kinase de type MB] et troponine), et l’angor instable (à gauche), dans lequel le segment ST reste isoélectrique. L’onde T correspond à la repolarisation des cellules myocardiques (après leur contraction). Elle est normalement positive (vers le haut). Elle s’inverse en cas de souffrance cellulaire (flèche courbe épaisse mauve). Le segment ST s’abaisse en dessous de la ligne isoélectrique lorsqu’il existe une souffrance étendue à l’ensemble de la couche la plus interne du myocarde (endocarde). Les antiagrégants limitent l’extension de la thrombose coronarienne, qui est partielle, et préviennent les emboles capillaires.
2. Physiopathologie des syndromes coronariens aigus
L’athérothrombose associe une lésion d’athérosclérose à une réaction thrombotique dans laquelle de nombreuses cellules sont impliquées. L’inflammation en est le dénominateur commun, et intervient à plusieurs étapes-clé de l’évolution de l’athérothrombose : activation de l’endothélium et recrutement des monocytes et des lymphocytes assurant la production locale et systémique des cytokines pro-inflammatoires, dégradation des protéines de la chape fibreuse par des protéases matricielles et, enfin, induction de l’apoptose des cellules de la plaque et formation du cœur lipidique procoagulant. La rupture ou l’érosion de la plaque résulte de la conjonction de sa fragilisation mécanique et des forces exercées sur elle (stimulus). Les plaques les plus vulnérables, et donc les plus à risque de rupture, sont celles comportant un centre lipidique occupant plus de 40 % du volume de la plaque et une chape fibreuse fine, riche en macrophages et pauvre en cellules musculaires lisses. L’histoire d’une plaque se résume à l’alternance entre des phases d’instabilité (liée à un processus inflammatoire) et des phases dites de cicatrisation (réparation de la chape). Lorsque la plaque est rompue, le déclenchement de la coagulation par les cellules inflammatoires aboutit à la thrombose, qui peut se lyser spontanément (thrombose/ thrombolyse : équilibre instable existant entre la thrombogenèse et la thrombolyse naturelle endogène), emboliser en totalité ou en partie dans la circulation d’aval, ou bien encore croître jusqu’à l’occlusion complète de la lumière artérielle. Lyse/remodelage indique que le thrombus résiduel qui n’a pu être dissous par la thrombolyse naturelle est incorporé dans la plaque, participant au processus complexe de sa croissance. Le thrombus s’organise alors et secondairement se recanalise. Dans tous les cas, il existe une cicatrisation de la plaque rompue avec incorporation du thrombus au sein de la plaque, permettant sa croissance. Lyse/réparation indique un mécanisme physiologique. Dans ce cas, le thrombus n’est pas pathologique mais sert à la réparation artérielle. Il ne s’étend pas et reste circonscrit au niveau de la zone de rupture. Il permet la réparation de l’endothélium ; c’est un processus naturel physiologique.
La première conséquence fonctionnelle majeure de l’activation des plaquettes est le changement de conformation des glycoprotéines GP IIb/IIIa présentes à leur surface et l’externalisation, dans leur conformation active, de nouvelles molécules de GP IIb/IIIa, ce qui aboutit à un nombre total d’environ 50 000 à 100 000 molécules par plaquettes. La GP IIb/IIIa activée interagit spécifiquement, en présence de calcium, avec plusieurs protéines circulantes, et notamment avec le fibrinogène, ce qui permet la formation de ponts entre les plaquettes activées (Figure 2) : c’est l’agrégation plaquettaire proprement dite.
 |
Figure 2. Interaction de la molécule de fibrinogène avec son récepteur membranaire plaquettaire activé, la glycoprotéine GP IIb/IIIa. Il s’agit de la voie finale commune de l’agrégation plaquettaire, sur laquelle joue les inhibiteurs de la GP IIb/IIIa. Les résidus RGD (Arg-Gly-Asp) de la molécule de fibrinogène, notamment ceux en position g400-411, sont des sites d’interaction spécifiques. |
La seconde conséquence fonctionnelle importante de l’activation des plaquettes est la libération du contenu de leurs granules. L’ADP et la sérotonine d’origine granulaire activent alors de nouvelles plaquettes adjacentes et participent à l’interaction paroi vasculaire/plaquettes. La synthèse du thromboxane A2 (TXA2) par les plaquettes activées à partir de l’acide arachidonique fournit un autre puissant agent vasoconstricteur favorisant l’agrégation plaquettaire (Figure 1).
La taille de l’agrégat plaquettaire dépend en grande partie des forces de cisaillement du flux vasculaire et de l’importance de la sténose artérielle [1]. Lorsque l’agrégat atteint une taille suffisante pour modifier les conditions locales d’écoulement du sang, la coagulation devient alors efficace : le réseau fibrillaire de fibrine stabilise l’agrégat plaquettaire. Le thrombus peut être intermittent, se fractionner et migrer sous forme d’emboles sous l’action des conditions de flux, ou au contraire être fixe et se développer pour occlure rapidement l’artère.
Les antiagrégants plaquettaires sont la pierre angulaire du traitement médicamenteux des SCA. On en distingue trois principaux en pratique clinique, dont les indications et les possibilités d’associations multiples compliquent les choix de stratégies thérapeutiques.
Inhibiteurs de l’activation plaquettaire
Aspirine
L’aspirine (acide acétylsalicylique, ASA) bloque la voie du thromboxane A2 en acétylant de façon irréversible la cyclooxygénase plaquettaire de type 1 (Figure 3). Elle doit être administrée aussi précocement que possible, sous la forme d’une dose de charge par voie intraveineuse (500 mg en injection unique) suivie d’une dose d’entretien par voie orale de 75 mg par jour. Son efficacité est immédiate, et son administration doit être poursuivie à vie [3]. Elle réduit de 47 % l’incidence combinée des décès et infarctus lorsqu’elle est administrée dès la phase aiguë des SCA [4, 5].
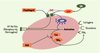 |
Figure 3. Principales voies d’activation plaquettaire menant à l’externalisation et à l’activation de la GP IIb/IIIa. La libération d’ADP par les globules rouges (GR) et les granules plaquettaires denses au site de la lésion vasculaire, de thromboxane A2 (TXA2) par les plaquettes activées, et surtout la production explosive de thrombine (par la cascade de la coagulation activée à la surface des plaquettes) permettent le recrutement et l’activation de nouvelles plaquettes, ce qui provoque un emballement du processus thrombotique au niveau de la lésion vasculaire. Le collagène du sous-endothélium est un agoniste plaquettaire puissant. Le TXA2, le collagène et la thrombine agissent chacun par l’intermédiaire d’un récepteur membranaire qui leur est spécifique. Le schéma montre également les mécanismes d’inhibition de l’agrégation plaquettaire par l’aspirine (ASA), sur la cyclooxygénase Cox et donc sur la production de TXA2, et par les thiénopyridines (clopidogrel), sur le récepteur de l’ADP. Ces deux antiagrégants limitent l’activation et l’externalisation du récepteur GP IIb/IIIa membranaire. |
Thiénopyridines
Les thiénopyridines (ticlopidine et clopidogrel) bloquent la voie de l’ADP - l’une des trois principales voies de l’activation plaquettaire par un mécanisme d’inhibition spécifique des récepteurs P1Y12 de l’ADP (Figure 3). Leur effet biologique est différé, car ces molécules administrées uniquement par voie orale doivent être métabolisées pour être actives. Une dose de charge est nécessaire pour obtenir un effet antiagrégant efficace plus rapidement. En raison de ses propriétés pharmacologiques et de sa meilleure tolérance digestive et hématologique que celle de la ticlopidine, le clopidogrel est devenu la thiénopyridine de référence. Deux grands essais thérapeutiques ont démontré son efficacité en association avec l’aspirine dans la maladie coronarienne.
L’étude randomisée CURE [6], menée auprès de 12 500 patients admis pour SCA sans élévation du segment ST, a montré que l’association de l’aspirine avec le clopidogrel (Plavix®) à la dose de 75 mg/j permettait de réduire de 20 % l’incidence des décès, infarctus ou accidents vasculaires cérébraux, par rapport à la combinaison aspirine-placebo (risque relatif = 0,80, p < 0,001). L’efficacité de ce traitement est observée dès la 48e heure et se maintient à long terme (un an). Le prix à payer est une augmentation des saignements majeurs (3,7 % contre 2,7 %) et le doublement des hémorragies dites mineures (essentiellement digestives). L’utilisation de faibles doses d’aspirine (75 mg/jour) permet de réduire le risque hémorragique de cette association. Il est donc actuellement recommandé d’administrer dès que possible le clopidogrel en combinaison avec l’aspirine, et de poursuivre cette association au moins neuf mois après un épisode de SCA sans élévation du segment ST. Toutefois, la dose de charge du clopidogrel, comme sa dose d’entretien, doivent encore être mieux déterminées.
L’étude CREDO [7], menée auprès de 2 116 patients, a confirmé les données de l’étude CURE, cette fois dans l’angioplastie coronarienne : une bithérapie antiagrégante aspirine-clopidogrel au long cours est supérieure à une monothérapie aspirine-placebo pour prévenir les complications ischémiques d’une angioplastie coronarienne. L’association de l’aspirine et du clopidogrel était jusqu’alors poursuivie un mois après une angioplastie, de façon à éviter les occlusions thrombotiques aiguës des endoprothèses coronariennes. Dans l’étude CREDO, un traitement prolongé par la combinaison aspirine-clopidogrel est associé à une diminution de 27 % de l’incidence des décès, infarctus du myocarde ou accidents vasculaires cérébraux par rapport à une monothérapie par aspirine (réduction du risque relatif = 26,9 % ; intervalle de confiance (IC) à 95 % = 3,9 % – 44,4 %, p = 0,02).
Inhibiteurs de l’agrégation plaquettaire : antagonistes de la GP IIb/IIIa
Ces molécules inhibent l’interaction du fibrinogène avec son récepteur plaquettaire, la glycoprotéine GP IIb/IIIa exprimée sur les plaquettes activées (Figure 2). On distingue l’anticorps monoclonal murin humanisé abciximab (Réopro) et les molécules de faible affinité ayant une vitesse de dissociation rapide, ou « petites molécules » (eptifibatide, tirofiban). L’abciximab bloque de façon non compétitive le récepteur IIb/IIIa, entraînant une modification de sa conformation. Il a la demi-vie la plus longue. Parmi les « petites molécules », on distingue l’eptifibatide (Integrilin), peptide de synthèse qui bloque de façon compétitive et réversible la liaison du fibrinogène au récepteur plaquettaire IIb/IIIa, et le tirofiban (Aggrastat), peptidomimétique de synthèse qui inhibe de façon compétitive la liaison fibrinogène-GPIIb/IIIa en mimant la charge et la conformation spatiale de la séquence RGD (Arg-Gly-Asp) du fibrinogène. Ces molécules sont administrées par voie intraveineuse pendant une courte durée, inférieure à 48 heures.
Prise en charge médicale des SCA sans élévation du segment ST
Les inhibiteurs de la GP IIb/IIIa n’apportent de bénéfice qu’aux patients à très haut risque (élévation de la troponine, patients diabétiques ou insuffisants rénaux, patients âgés, patients instables en attente de pontage) [8]. En effet, cette classe thérapeutique ne réduit que de 10 % le risque de décès ou d’infarctus chez les patients admis pour SCA sans élévation du ST (10,8 % contre 11,8 % ; odds ratio (OR) = 0,91 ; IC à 95 % : 0,84 – 0,98, p = 0,015, pour placebo et inhibiteurs des IIb/IIIa, respectivement). En revanche, cette réduction atteint 15 % chez les patients ayant une troponine positive (10,3 % contre 12,0 % ; OR = 0,85 ; IC à 95 % = 0,71 – 1,03).
Utilisation au cours de l’angioplastie des SCA
L’angioplastie coronarienne, qui consiste à détruire le thrombus et la plaque sous-jacente, ainsi que le stent (‹), qui permet le maintien d’un calibre artériel suffisant, ont un effet prothrombogène bien démontré.
(‹) m/s 2004, n° 1, p. 98
Les inhibiteurs de la GP IIb/IIIa évitent les complications de l’angioplastie. Ainsi, la réduction des décès, infarctus et thrombose nécessitant une revascularisation urgente de l’artère traitée atteint 50 % en cas de traitement conjoint par un inhibiteur de la GP IIb/IIIa (8,0 % contre 4,9 % pour le placebo, p = 0,0001) [9, 10, 24–27]. Il existe, de plus, un effet synergique de l’association endoprothèse/anti-IIb/IIIa.
Par ailleurs, l’administration précoce d’inhibiteurs de la GP IIb/IIIa à la phase aiguë d’un infarctus du myocarde permet de réouvrir l’artère coronaire occluse avant d’effectuer l’angioplastie, avec un taux de succès proportionnel au délai d’administration du médicament par rapport au début des symptômes : ainsi, l’administration préhospitalière d’abciximab multiplie par trois la fréquence de revascularisation de l’artère coronaire ouverte [11, 28].
Prise en charge des infarctus traités par thrombolyse
L’association d’un inhibiteur de la GP IIb/IIIa avec une demi-dose des agents thrombolytiques rt-PA (activateur tissulaire du plasminogène) ou TNK (ténecteplase) réduit de 50 % l’incidence des rechutes d’infarctus par rapport à la thrombolyse seule. Cependant, en raison d’une augmentation très significative des complications hémorragiques majeures, en particulier des hémorragies intracérébrales [12, 13], l’association d’un inhibiteur de la GP IIb/IIIa avec la thrombolyse n’est pas à recommander en première intention dans cette indication. L’avenir est d’en évaluer l’intérêt dans des stratégies de reperfusion dites combinées, où seront comparées l’angioplastie primaire en association avec les inhibiteurs de la GP IIb/IIIa et l’angioplastie facilitée par thrombolyse (demi-dose de thrombolytiques + inhibiteurs de la GP IIb/IIIa, ou thrombolyse seule à pleine dose).
Complications hémorragiques liées aux inhibiteurs de la GP IIb/IIIa
Les anti-GPIIb/IIIa favorisent systématiquement les complications hémorragiques majeures par rapport au placebo chez les patients admis pour SCA sans élévation du segment ST (2,4 % contre 1,4 %, OR = 1,62 ; IC à 95 % = 1,36-1,94, p < 0,0001) [8]. En revanche, aucune augmentation de l’incidence des hémorragies cérébrales (0,08 % chez l’ensemble des patients) n’a été mise en évidence à ce jour.
Le choix des associations thérapeutiques est ici important. Les anticoagulants (héparine non fractionnée et héparines de bas poids moléculaire) sont, en association avec l’aspirine, la pierre angulaire des traitements antithrombotiques utilisés dans les SCA. En effet, l’utilisation des héparines de bas poids moléculaire à la place de l’héparine standard dans les SCA réduit de moitié le risque d’hémorragie majeure liée à l’utilisation des anti-GP IIb/IIIa [14, 15].
Faut-il systématiquement associer le clopidogrel et les inhibiteurs de la GP IIb/IIIa ?
Cette question trouve sa justification dans le risque hémorragique et le coût liés aux traitements par les inhibiteurs de la GP IIb/IIIa.
D’une part, l’étude PEACE montre que les anti-IIb/IIIa ont un effet antiagrégant s’ajoutant à celui de l’aspirine et du clopidogrel [16]. Elle suggère aussi qu’un traitement prolongé par anti-GP IIb/IIIa (en particulier par les petites molécules) semble avoir un effet agoniste partiel, comme le confirme l’augmentation de l’expression de la P-sélectine à la surface de la membrane plaquettaire. Ces données, déjà rapportées avec les formes orales d’inhibiteurs de la GP IIb/IIIa, plaident en faveur de traitements de courte durée (< 48 heures). D’autre part, tous les essais cliniques montrent que le clopidogrel n’atténue pas le bénéfice clinique des anti-GP IIb/IIIa dans la prévention des complications de l’angioplastie coronarienne [7, 9, 17, 18]. En particulier, l’étude CREDO, dans laquelle les patients ont été prétraités par le clopidogrel avant l’angioplastie, montre une réduction des événements ischémiques de 45,3 % dans le sous-groupe traité par une trithérapie antiagrégante, contre 28,3 % dans le sous-groupe de patients ne recevant pas d’anti-GP IIb/IIIa.
Il faut retenir qu’à l’inverse de l’association aspirine/clopidogrel, qui est efficace sur le long cours quels que soient la stratégie thérapeutique (traitement invasif ou médical conservateur) utilisée et le profil de risque des patients, la trithérapie antiagrégante associant aspirine, clopidogrel et anti-GP IIb/IIIa est surtout efficace lorsqu’une angioplastie est effectuée dans un contexte de SCA. Cette classe thérapeutique permet alors de réduire de 50 % les complications thrombotiques aiguës liées au geste d’angioplastie, ainsi que la mortalité à long terme (à trois ans).
Conclusions et perspectives
Si l’efficacité des antiagrégants dans le traitement des syndromes coronariens aigus est clairement établie, leur modalité d’utilisation est perfectible. En effet, les recommandations des sociétés savantes et les autorisations de mise sur le marché des molécules ne répondent pas complètement aux attentes des praticiens. Trois défis sont à relever.
Il faut ainsi mieux évaluer l’intérêt de ces antiagrégants plaquettaires chez les patients exclus des essais randomisés, qui représentent environ 50 % des patients tout-venant [19]. Ces patients, souvent âgés, insuffisants rénaux ou insuffisants cardiaques, ont un risque plus élevé de complications ischémiques comme hémorragiques. En particulier, la sécurité d’utilisation au long cours d’une association d’antiagrégants oraux doit impérativement être évaluée chez eux. Il reste également à développer et valider des outils biologiques de surveillance, afin de définir les posologies optimales, en particulier avec les inhibiteurs de la GP IIb/IIIa, qui sont particulièrement coûteux et exposent à des risques de complications hémorragiques [20, 21]. Ces mêmes outils devraient aussi permettre de dépister les résistances biologiques aux antiagrégants plaquettaires oraux, afin de mieux cerner les patients « mauvais répondeurs ». Enfin, l’arrêt temporaire de ces traitements prescrits au long cours est un sujet de controverse récurrent entre les praticiens de différentes spécialités, cardiologues, anesthésistes ou chirurgiens. Pourtant, il s’agit d’une situation clinique très fréquente, très mal évaluée et source de récidive d’accidents coronariens aigus [22, 23], qu’il est important d’étudier en détail.
Le bénéfice clinique des associations d’antiagrégants plaquettaires confirme les données les plus récentes sur le rôle physiopathologique des plaquettes dans les complications aiguës de la maladie athérothrombotique coronarienne. Ce bénéfice est encore plus grand lorsqu’un traitement par angioplastie y est associé, inducteur d’une instabilité supplémentaire de la paroi artérielle.
Références
- Davies SW, Marchant B, Lyons JP. Irregular coronary lesion morphology after thrombolyis predicts early clinical instability. J Am Coll Cardiol 1991; 18 : 669–74. [Google Scholar]
- Theroux P, Fuster V. Acute coronary syndromes. Unstable angina and non-Q-wave myocardial infarction. Circulation 1998; 97 : 1195–206. [Google Scholar]
- Braunwald E, Antman EM, Califf RM, et al. ACC/AHA Guideline update for the management of patients with unstable angina and non-ST-segment elevation myocardial infarction, 2002. Summary article : a report of the American college of cardiology/American heart association task force on practice guidelines (committee on the management of patients with unstable angina). Circulation 2002; 106 : 1893–900. [Google Scholar]
- Verstaete M. A European view on the North American fifth consensus on antithrombotic therapy. Chest 2000; 117 : 1755–70. [Google Scholar]
- ISIS-2 collaborative group. Randomized trial of IV streptokinase, oral aspirin, both or neither among 17187 cases of suspected acute myocardial infarction. Lancet 1988; 330 : 1287–94. [Google Scholar]
- The clopidogrel in unstable angina to prevent recurrent events trials investigators (CURE). Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345 : 494–502. [Google Scholar]
- Steinhubl SR, Berger S, Mann JT, et al. Early and sustained dual oral antiplatelet therapy following percutaneous coronary intervention. JAMA 2002; 288 : 2411. [Google Scholar]
- Boersma E, Harrington RA, Moliterno DJ, et al. Platelet glyco-protein IIb/IIIa inhibitors in acute coronary syndromes : a meta-analysis of all major randomised clinical trials. Lancet 2002; 359 : 189–98. [Google Scholar]
- The EPISTENT investigators. Randomised placebo-controlled and balloon-angioplasty-controlled trial to assess safety of coronary stenting with use of platelet glycoprotein IIb/IIIa blockade. Lancet 1998; 352 : 87–92. [Google Scholar]
- The ESPRIT investigators. Novel dosing regimen of eptifibatide in planned coronary stent implantation (ESPRIT) : a randomised, placebo-controlled trial. Lancet 2000; 356 : 2037–44. [Google Scholar]
- Montalescot G, Barragan P, Wittenberg O, et al. Platelet glycoprotein IIb/IIIa inhibition with coronary stenting for acute myocardial infarction. N Engl J Med 2001; 344 : 1895–903. [Google Scholar]
- The GUSTO V investigators. Reperfusion therapy for acute myocardial infarction with fibrinolytic therapy or combination reduced fibrinolytic therapy and platelet glycoprotein IIb/IIIa inhibition : the GUSTO V randomised trial. Lancet 2001; 357 : 1905–14. [Google Scholar]
- The Assessment of the safety and efficacy of a new thrombolytic regimen (ASSENT)-3 investigators. Efficacy and safety of tenecteplase in combination with enoxaparin, abciximab or unfractionated heparin : the ASSENT-3 randomised trial in acute myocardial infarction. Lancet 2001; 358 : 605–13. [Google Scholar]
- Cohen M, Theroux P, Borzak S, et al. Randomized double-blind safety study of enoxaparin versus unfractionated heparin in patients with non-ST-segment elevation acute coronary syndromes treated with tirofiban and aspirin : the ACUTE (antithrombotic combination using tirofiban and enoxaparin) II study. Am Heart J 2002; 144 : 470–7. [Google Scholar]
- Goodman SG, Fitchett D, Armstrong PW, et al. Randomized evaluation of the safety and efficacy of enoxaparin versus unfractionated heparin in high-risk patients with non-ST-segment elevation acute coronary syndromes receiving the glycoprotein IIb/IIIa inhibitor eptifibatide. Circulation 2003; 107 : 238–44. [Google Scholar]
- Dalby M, Montalescot G, Sollier CB, et al. Eptifibatide provides additional platelet inhibition in non-ST-elevation myocardial infarction patients already treated with aspirin and clopidogrel. Results of the platelet activity extinction in non-Q-wave myocardial infarction with aspirin, clopidogrel, and eptifibatide (PEACE) study. J Am Coll Cardiol 2004; 43 : 162–8. [Google Scholar]
- Cannon CP, Weintraub SW, Demopoulos L, et al. A comparison of invasive versus conservative strategy in patients with unstablecoronary syndromes treated with the glycoprotein IIb/IIIa inhibitor tirofiban. N Engl J Med 2001; 344 : 1879–87. [Google Scholar]
- Topol EJ, Moliterno DJ, Herrmann HC, et al. Comparison of two platelet glycoprotein IIb/IIIa inhibitors, tirofiban and abciximab, for the prevention of ischemic events with percutaneous coronary revascularization. N Engl J Med 2001; 344 : 1888–94. [Google Scholar]
- Collet JP, Montalescot G, Fine E, et al. Enoxaparin in unstable angina patients who would have been excluded from randomized pivotal trials. J Am Coll Cardiol 2003; 41 : 8–14. [Google Scholar]
- Sharma SK, Kini AS, Perez N, et al. Tirofiban, eptifibatide and abciximab in minimizing myocardial damage during high-risk coronary intervention : final results of the TEAM pilot trial. J Am Coll Cardiol 2002; 39 (suppl A) : 73A (abstract 887-5). [Google Scholar]
- Gum PA, Kottle-Marchant K, Welch PA, et al. A prospective, blinded determination of the natural history of aspirin resistance among stable patients with cardiovascular disease. J Am Coll Cardiol 2003; 41 : 961–5. [Google Scholar]
- Collet JP, Himbert D, Steg PG. Myocardial infarction after aspirin cessation in stable coronary artery disease patients. Int J Cardiol 2000; 76 : 257–8. [Google Scholar]
- Samama CM, Bastien O, Forestier F, et al. Antiplatelet agents in the perioperative period : expert recommendations of the French society of anesthesiology and intensive care (SFAR) 2001. Summary statement. Can J Anaesth 2002; 49 : S26–35. [Google Scholar]
- The EPILOG investigators. Platelets glycoprotein IIb/IIIa receptor blockade and low-dose heparin during percutaneous coronary revascularzation. N Engl J Med 1997; 336 : 1689–96. [Google Scholar]
- The Epic investigators. Use of a monoclonal antibody directed against the platelet glycoprotein IIb IIIa receptor in high risk coronary angioplasty. N Engl J Med 1994; 330 : 956–61. [Google Scholar]
- The CAPTURE investigators. Randomized placebo-controlled trial of abciximab before and during coronary intervention in refractory unstable angina : the CAPTURE study. Lancet 1997; 349 : 1429–35. [Google Scholar]
- Brener SJ, Barr LA, Burchenal JE, et al. Randomized, placebo-controlled trial of platelet glycoprotein IIb/IIIa blockade with primary angioplasty for acute myocardial infarction. ReoPro and primary PTCA organization and randomized trial (RAPPORT) investigators. Circulation 1998; 98 : 34–41. [Google Scholar]
- Stone GW, Grines CL, Cox DA, et al. Comparison of angioplasty with stenting, with or without abciximab, in acute myocardial infarction. N Engl J Med 2002; 346 : 957–66. [Google Scholar]
Liste des figures
|
Figure 1. Formation de l’agrégat plaquettaire et du réseau de fibrine à la surface de la plaque rompue. Les plaquettes forment dans un premier temps un tapis au niveau de la plaque lésée. L’interaction de la glycoprotéine Iba (GP Ib) plaquettaire avec le facteur von Willebrand (FvW) est primordiale dans des conditions de contraintes de cisaillement élevées, par exemple en cas de sténose athéromateuse serrée dans un vaisseau de petit diamètre : elle permet en effet l’adhérence des plaquettes au sous-endothélium activé. L’activation plaquettaire survient dans un deuxième temps, et résulte soit de l’adhérence des plaquettes au sous-endothélium, soit de leur exposition à des agonistes circulants, notamment la thrombine et le collagène, premiers activateurs trouvés au niveau de la brèche vasculaire. La présence de facteur tissulaire (FT) dans des microparticules libérées par les cellules inflammatoires de la plaque entraîne l’activation de la coagulation. Le complexe Xa/Va (prothrombinase) permet la transformation de la prothrombine en thrombine (IIa) (le facteur VIIIa est un puissant cofacteur de la production de thrombine), à l’origine de la formation de fibrine à partir du fibrinogène. Le complexe GP IIb/IIIa est quant à lui responsable de l’agrégation plaquettaire par le fibrinogène, qui forme des ponts interplaquettaires (voir Figure 2). La sérotonine, le thromboxane A2 (TXA2) et l’ADP sont libérés par les plaquettes pour activer et recruter de nouvelles plaquettes. |
| Dans le texte | |
|
Figure 2. Interaction de la molécule de fibrinogène avec son récepteur membranaire plaquettaire activé, la glycoprotéine GP IIb/IIIa. Il s’agit de la voie finale commune de l’agrégation plaquettaire, sur laquelle joue les inhibiteurs de la GP IIb/IIIa. Les résidus RGD (Arg-Gly-Asp) de la molécule de fibrinogène, notamment ceux en position g400-411, sont des sites d’interaction spécifiques. |
| Dans le texte | |
|
Figure 3. Principales voies d’activation plaquettaire menant à l’externalisation et à l’activation de la GP IIb/IIIa. La libération d’ADP par les globules rouges (GR) et les granules plaquettaires denses au site de la lésion vasculaire, de thromboxane A2 (TXA2) par les plaquettes activées, et surtout la production explosive de thrombine (par la cascade de la coagulation activée à la surface des plaquettes) permettent le recrutement et l’activation de nouvelles plaquettes, ce qui provoque un emballement du processus thrombotique au niveau de la lésion vasculaire. Le collagène du sous-endothélium est un agoniste plaquettaire puissant. Le TXA2, le collagène et la thrombine agissent chacun par l’intermédiaire d’un récepteur membranaire qui leur est spécifique. Le schéma montre également les mécanismes d’inhibition de l’agrégation plaquettaire par l’aspirine (ASA), sur la cyclooxygénase Cox et donc sur la production de TXA2, et par les thiénopyridines (clopidogrel), sur le récepteur de l’ADP. Ces deux antiagrégants limitent l’activation et l’externalisation du récepteur GP IIb/IIIa membranaire. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.