")
")
| Issue |
Med Sci (Paris)
Volume 20, Number 2, Février 2004
|
|
|---|---|---|
| Page(s) | 175 - 181 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/2004202175 | |
| Published online | 15 February 2004 | |
Rôle de l’immunité dans l’athérosclérose et dans les syndromes coronariens aigus
Role of the immune response in atherosclerosis and acute coronary syndromes
1
Inserm EMI 00-16, Faculté de médecine Necker-Enfants Malades, 156, rue de Vaugirard, 75015 Paris, France
2
Service de cardiologie, Hôpital Européen Georges Pompidou, 20, rue Leblanc, 75015 Paris, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
La formation et le développement des plaques d’athérome résultent d’une interaction dynamique entre la paroi des vaisseaux et le sang circulant. L’étape cruciale de la constitution des plaques semble être la présence de lipoprotéines oxydées dans l’espace sous-endothélial, qui entraîne l’expression locale de facteurs responsables de l’infiltration de la paroi par des monocytes et des lymphocytes T. L’athérosclérose est ainsi une maladie immuno-inflammatoire, différents composants du système immunitaire assumant un rôle protecteur ou délétère dans le devenir de la maladie. D’où l’idée de développer, à partir de la modulation de la réponse immunitaire, de nouvelles thérapeutiques préventives et curatives pour lutter contre les maladies cardiovasculaires, première cause de décès dans les pays industrialisés.
Abstract
Cardiovascular diseases represent one of the most important causes of death in the world. The underlying pathogenetic process is atherosclerosis which leads to the progressive reduction of the arterial lumen and therefore to the ischemia of the perfused organs. Atherogenesis results from the interaction between the biology of the arterial wall and the various stress stimuli present in the circulating blood. The first steps of atherogenesis occur very early, already during the fetal life. Those arterial segments that are subjected to the initiating causes (including hemodynamic stress) show altered endothelial permeability and allow the infiltration of macromolecules, like lipoproteins, in the subintimal space. At the same time, the smooth muscle cells that are subjected to the same local factors produce proteoglycans able to bind lipoproteins and to promote their oxidation. Oxidized lipoproteins induce the expression of chemokines and adhesion molecules on the luminal surface of the endothelium, which then allow the local recruitment of monocytes-macrophages and T lymphocytes. This is a local inflammatory process that, in theory, should contribute to reestablish the homeostasis of the vascular wall by promoting the elimination of injured tissue and its repair. Unfortunately, for unknown reasons, the immuno-inflammatory reaction persists and autoamplifies, the various components of the immune response finally contributing to the pathogenesis of atherosclerosis as well as of atherosclerotic complications.
© 2004 médecine/sciences - Inserm / SRMS
Les maladies cardiovasculaires représentent l’une des principales causes de mortalité dans le monde [1]. La cause sous-jacente de cette mortalité élevée est le processus athéroscléreux qui est responsable du rétrécissement progressif de la lumière des vaisseaux (sténose) et entraîne l’ischémie des organes distaux. Les premières étapes de l’athérogenèse ont lieu très précocement, dès la vie fœtale [2]. Dans les régions sujettes aux lésions, en relation avec des facteurs hémodynamiques locaux, la perméabilité endothéliale augmentée conduit à une infiltration de macromolécules, comme les lipoprotéines, dans l’intima. Dans le même temps, les cellules musculaires lisses, soumises aux mêmes facteurs hémodynamiques locaux, produisent des protéoglycanes capables de se lier aux lipides, et de faciliter ainsi leur oxydation [3]. L’endothélium, sollicité par les lipoprotéines oxydées, exprime des chimiokines et des molécules d’adhérence, ce qui permet l’infiltration locale des monocytes et des lymphocytes T [4]. S’amorce alors un processus immuno-inflammatoire local qui, théoriquement, devrait prendre en charge la destruction et le « nettoyage », mais aussi organiser la réparation de la paroi vasculaire. Or, pour des raisons non encore complètement élucidées, l’issue de ce processus immuno-inflammatoire n’est pas optimale, et les différents composants de la réponse immune peuvent en fait participer à la pathogénie de l’athérosclérose tout au long de son évolution [5].
Origine de l’hypothèse inflammatoire de l’athérosclérose
La présence de cellules inflammatoires dans les plaques d’athérome est connue depuis le xixe siècle [6]. Mais ce n’est qu’au début des années 1990 que les chercheurs ont manifesté un intérêt majeur pour le rôle de l’inflammation dans la pathogénie de l’athérosclérose. En 1985, C.J. Schwartz émet l’hypothèse selon laquelle l’athérosclérose est une maladie inflammatoire de la paroi artérielle impliquant la participation active des monocytes-macrophages [7]. La même année, K. Kohchi montre que les artères coronaires responsables d’angor instable sont particulièrement infiltrées par des cellules inflammatoires, qui sont surtout regroupées dans la couche adventitielle autour des terminaisons nerveuses et peuvent donc participer à la composante dynamique (spasme) de l’instabilité clinique [8].
Inflammation et formation des plaques d’athérome : faits et hypothèses
La phase la plus précoce de formation de la plaque est marquée par l’infiltration de leucocytes dans la paroi artérielle. L’extravasation des lipoprotéines de basse densité (LDL) et leur oxydation dans l’espace sous-endothélial pourraient constituer l’étape cruciale dans la formation de la plaque [2]. Les LDL oxydées (oxLDL) induisent l’expression de molécules d’adhérence [9] et de chimiokines [10], ouvrant ainsi aux leucocytes l’accès à la paroi vasculaire (Figure 1). Les macrophages internalisent les oxLDL par un mécanisme non soumis à régulation : en conséquence, la quantité d’oxLDL accumulée à l’intérieur des macrophages dépasse la capacité de ces derniers à les dégrader. Ces macrophages se transforment alors en cellules spumeuses, qui demeurent dans l’intima et constituent les premières lésions d’athérome.
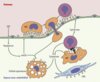 |
Figure 1. L’hypothèse inflammatoire de l’athérogenèse. Dans l’hypothèse la plus accréditée aujourd’hui, l’extravasation des lipoprotéines de basse densité (LDL) et leur oxydation dans l’espace sous-endothélial représente la première étape de l’athérogenèse. Les LDL oxydées (oxLDL) induisent l’expression de molécules d’adhérence et de récepteurs de chimiokines à la surface des cellules endothéliales (CE), ouvrant ainsi aux monocytes (MO) l’accès à la paroi vasculaire. Une fois entrés, les monocytes-macrophages internalisent les oxLDL, les accumulent à l’intérieur et se transforment ainsi en cellules spumeuses. Des lymphocytes T (LT) sont également recrutés, probablement alertés par les cytokines sécrétées par les macrophages (MP) et par des molécules d’adhérence et des chimiokines exprimées par les cellules endothéliales et musculaires lisses (CML) activées. Ces lymphocytes sécrètent des cytokines et expriment des marqueurs d’activation, qui signent vraisemblablement une stimulation antigénique locale, pouvant entraîner des effets importants sur le devenir de plaques précoces, ainsi que sur la biologie de la paroi artérielle et la pathogénie des complications des plaques (d’après [5]). |
La composante cellulaire des plaques comprend également un grand nombre de lymphocytes, probablement alertés par des cytokines et des chimiokines produites par les macrophages et par les cellules endothéliales et musculaires lisses activées. Ces lymphocytes expriment des marqueurs d’activation et reconnaissent des épitopes contenus dans les oxLDL [11]. Les peptides antigéniques dérivés des oxLDL peuvent être présentés aux lymphocytes T par les macrophages locaux [12]. Ainsi, dès les stades précoces de l’athérogenèse, les lésions contiennent tous les composants d’une réponse immunitaire spécifique locale. Une réponse immunitaire spécifique aux oxLDL est également détectable dans la rate de souris hypercholestérolémiques âgées, mais pas chez des souris jeunes [13]. Il est par conséquent possible que, avec la progression de la maladie, la réponse initiale immunitaire locale s’étende aux organes lymphoïdes secondaires.
Inflammation et athérosclérose coronaire
La principale manifestation clinique des lésions coronaires d’athérosclérose provient soit d’une occlusion lente et progressive de la lumière, due à une augmentation de taille de la plaque (angor stable), soit à une sténose aiguë, due à la formation soudaine d’un thrombus associé ou non à une vasoconstriction (syndromes coronaires aigus : angor instable ou infarctus du myocarde). L’angor stable est associé à un état inflammatoire de faible intensité, et chronique, localement (dans les plaques) comme en périphérie (sang circulant).
Le développement des syndromes coronaires aigus repose sur la transformation des plaques stables « quiescentes » en plaques instables, accompagnées de la formation d’un thrombus et responsables d’une ischémie soudaine (lésions coupables). L’étude des lésions coupables révèle souvent une érosion ou une rupture de la plaque [14, 15]. Les matériaux procoagulants présents dans la matrice extracellulaire de la chape fibreuse, dans le cœur lipidique ou exprimés par les macrophages infiltrés dans les plaques seraient alors exposés à la circulation et pourraient déclencher une activation du système de la coagulation conduisant à l’occlusion aiguë par un thrombus. Étant donné qu’une inflammation aiguë et de forte intensité, à la fois dans les plaques et dans le sang, s’accompagne de phases d’instabilité clinique, il est possible que les composants de la réponse immuno-inflammatoire jouent un rôle important dans la précipitation des syndromes coronariens aigus.
En 1974, déjà, G.D. Friedman avait montré que le nombre de leucocytes circulants était un facteur de risque indépendant d’infarctus du myocarde [16]. À partir de 1982, de nombreuses études cliniques ont démontré la présence de marqueurs d’inflammation systémique, dont la protéine C réactive (CRP, C-reactive protein) [17], dans le sang des patients souffrant d’angor instable ou d’infarctus du myocarde. Il est possible que ces marqueurs jouent également un rôle direct dans la pathogénie de syndromes coronariens aigus (Tableau I).
Rôle possible de marqueur d’inflammation dans l’athérosclérose coronarienne. CRP : protéine C réactive ; SAA : serum amyloid A protein ; CE : cellules endothéliales ; CML : cellules musculaires lisses ; MO : monocytes ; LDL : lipoprotéines de basse densité ; IL : interleukine ; IFN : interféron.
Dans la mesure où les lymphocytes T infiltrés dans les plaques instables [18], de même que les lymphocytes T circulants [19], sont activés de façon transitoire (limitée aux phases d’instabilité clinique), nous avons formulé l’hypothèse selon laquelle l’inflammation faisait partie, dans l’angor instable, d’une réaction du système immunitaire. En réalité, l’angor instable qui répond au traitement est associé à une réponse immunitaire retardée, passagère et non inflammatoire (concentrations basses de CRP) [20], tandis que, à l’opposé, la persistance de l’instabilité clinique, malgré le traitement vasodilatateur et antithrombotique, est caractérisée par une activation précoce des lymphocytes T accompagnée d’une élévation des concentrations de CRP [19, 21]. Cela renforce l’hypothèse selon laquelle une réponse immunitaire de type inflammatoire pourrait nuire à l’évolution des syndromes coronariens aigus.
L’activation du système immunitaire peu de temps après la survenue des symptômes dans l’angor instable suggère que l’instabilité pourrait être déclenchée par des stimulus antigéniques (Figure 2). Une réponse T spécifique pour un antigène est caractérisée par l’expansion de lymphocytes portant un récepteur spécifique (TCR, T-cell receptor) reconnaissant l’antigène en question. L’analyse du répertoire du TCR renseigne donc sur l’origine monoclonale des lymphocytes. Dans l’angor instable, les cellules T activées ont un répertoire TCR restreint, contrairement aux patients atteints d’angor stable qui présentent un répertoire polyclonal normal [22], suggérant que la réponse T est spécifique pour un nombre limité d’antigènes seulement dans les cas d’instabilité clinique, alors que la simple présence d’athérome coronaire (angor stable) n’entraîne pas de réponse spécifique dépendante de l’antigène.
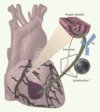 |
Figure 2. ’hypothèse inflammatoire dans les syndromes coronariens aigus. Une réponse immuno-inflammatoire aiguë et transitoire est détectable dans le sang circulant des patients présentant des syndromes coronariens aigus. L’activation des lymphocytes T et B (production d’anticorps, Ac) suggère que l’instabilité pourrait avoir été déclenchée par des stimulus antigéniques (comme l’indique la présence de pics monoclonaux dans l’analyse de l’ARNm codant pour le récepteur de l’antigène des lymphocytes T activés, par la technique de l’immunoscope), et que la réponse immuno-inflammatoire associée pourrait participer directement à la pathogénie de l’instabilité. Cette réponse immuno-inflammatoire devient détectable peu de temps après la survenue des symptômes dans l’angor instable. En fait, certains antigènes cibles contenus dans les plaques ne deviendraient accessibles aux lymphocytes circulants que lors d’une complication telle que la rupture de la chape fibreuse. Cela pourrait indiquer que la réponse immuno-inflammatoire observée dans les phases d’instabilité de l’athérosclérose coronaire pourrait être une conséquence, plutôt qu’une cause, de l’instabilité clinique. Dans ce cas, il reste plausible que la réponse immuno-inflammatoire joue un rôle important dans l’issue de l’instabilité clinique. |
Les antigènes candidats dans l’angor instable pourraient être localisés dans les plaques compliquées. À partir des endoathérectomies coronariennes, nous avons recherché la présence de cibles antigéniques candidates parmi les protéines des lésions coupables. La prolifération lymphocytaire en réponse aux extraits protéiques des lésions a tendance à être plus élevée chez les patients atteints d’angor instable que chez les patients stables [22]. Ce résultat est compatible avec la notion selon laquelle la plaque instable peut représenter une cible de la réponse immunitaire spécifique. Les oxLDL présentes dans les plaques d’athérosclérose [23] peuvent représenter une cible importante pour plusieurs raisons : elles sont reconnues par les lymphocytes T infiltrant les plaques [11], des concentrations élevées d’immunoglobulines circulantes spécifiques des oxLDL sont une caractéristique des patients présentant une athérosclérose sévère [24] et, enfin, les lymphocytes T circulants de patients souffrant d’angor instable réfractaire prolifèrent in vitro en réponse aux oxLDL [22]. Étant donné que les oxLDL peuvent être exposées dans les lésions coupables durant le processus d’instabilité, il est possible qu’une réponse immunitaire inflammatoire dirigée à leur encontre puisse prévenir la réparation tissulaire et aggraver ainsi les dommages dans les lésions coupables. En effet, les oxLDL présentes dans les plaques [23] peuvent induire la production d’interleukine 12 (IL-12) par les macrophages, et donc déclencher une réponse immunitaire Th1 [25] qui est supposée nuisible dans l’athérosclérose (voir ci-dessous).
Rôle des différents composants de la réponse immuno-inflammatoire
Polynucléaires neutrophiles
Les polynucléaires neutrophiles figurent parmi les premières cellules impliquées dans les réponses inflammatoires aux sites de lésions tissulaires, et il est vraisemblable qu’ils prennent également part au processus athéroscléreux, surtout pendant les phases d’instabilité clinique où l’inflammation devient détectable dans le sang circulant. En fait, la corrélation statistique entre le nombre de leucocytes circulants et le risque d’infarctus du myocarde [16] est surtout due aux neutrophiles. Il a été montré que les neutrophiles s’accumulent dans les plaques compliquées (c’est-à-dire dans les lésions où la surface interne du vaisseau est endommagée) des patients présentant des syndromes coronariens aigus [26]. Il faut signaler que, chez ces patients, les neutrophiles circulants sont également activés [27], et que leur activation est renforcée lors de leur passage à travers les artères athéroscléreuses non coupables [28].
Macrophages
Mis à part leur rôle de cellules spumeuses et présentatrices d’antigènes, les macrophages peuvent contribuer directement à la progression des lésions, par l’intermédiaire de l’oxydation des lipoprotéines, d’une part, et de la production de facteurs de croissance, agissant sur la prolifération des cellules musculaires lisses, et de métalloprotéases, capables de dégrader la matrice extracellulaire, d’autre part [5]. Ces effets sont supposés être néfastes : la dégradation du collagène pourrait être à la base de la vulnérabilité des plaques en facilitant leur rupture. Il faut néanmoins prendre en compte le fait que la dégradation de la matrice extracellulaire est une étape précoce et fondamentale dans le processus de réparation tissulaire. Si l’on considère que les macrophages peuvent faciliter la prolifération des cellules musculaires lisses, il est donc également plausible que les macrophages jouent un rôle bénéfique dans la réparation et la stabilisation des plaques compliquées.
Lymphocytes T
Les lymphocytes T peuvent aussi jouer un rôle à la fois protecteur et nuisible : les cellules pro-inflammatoires (Th1) contribuent à la pathogénie des maladies auto-immunes spécifiques d’organe, alors que les cellules anti-inflammatoires (Th2) les préviennent [29]. Dans l’athérome, les macrophages produisent et libèrent de l’IL-12, un puissant promoteur de la voie de différenciation Th1, et les cellules endothéliales des plaques expriment la P- et la E-sélectine qui recrutent préférentiellement les lymphocytes Th1 [30]. Chez les sujets atteints d’angor instable, les lymphocytes T activés produisent surtout de l’IFN-γ [31], une cytokine de la voie Th1. La libération d’IFN-γ par les cellules Th1 peut avoir des effets complexes sur l’évolution de l’athérosclérose. Tout d’abord, l’IFN-γ inhibe la synthèse du collagène et peut ainsi contribuer à diminuer la résistance de la chape fibreuse et favoriser une rupture de la plaque. Par ailleurs, il supprime la production de métalloprotéases induite par le TNF-α et l’IL-1β [32], cette action pouvant prévenir la dégradation de la matrice extracellulaire. La réponse des cellules T peut aussi déclencher la thrombose sur plaque, en favorisant l’expression de facteurs tissulaires par les macrophages et en inhibant la fonction anticoagulante de l’endothélium. Les souris athéroscléreuses déficientes en IL-10 (une cytokine de la voie Th2) montrent une exagération de la réponse Th1 ; les lésions avancées de ces souris sont enrichies en métalloprotéases et en facteurs procoagulants [33]. À l’opposé, la réduction de l’IFN-γ (et donc de la réponse Th1) par approche pharmacologique entraîne une réponse davantage de type Th2, qui est associée à une réduction de la taille des lésions avancées [34].
Anticorps
Outre la réponse immunitaire cellulaire, l’athérosclérose est également associée à une activation globale de l’immunité à médiation humorale [35]. Parmi les nombreux anticorps spécifiques décrits dans la littérature, une élévation du titre d’anticorps dirigés contre les oxLDL est caractéristique des patients athéroscléreux [24]. Ces anticorps peuvent former des complexes avec les oxLDL circulantes et activer la cascade du complément, contribuant ainsi aux dommages infligés au vaisseau. Néanmoins, ces complexes peuvent également favoriser l’élimination des oxLDL par les monocytes circulants et par les macrophages présents dans les plaques d’athérome ou en périphérie (notamment dans la rate). Que des titres réduits d’anticorps anti-oxLDL après splénectomie soient associés à une augmentation marquée de la taille de la lésion des souris déficientes en apolipoprotéine E [36] renforce cette hypothèse. De plus, l’immunisation des souris athéroscléreuses avec des oxLDL augmente le titre d’anticorps spécifiques et conduit à une réduction de la taille des lésions chez la souris [37, 38].
Lymphocytes B
Dans les modèles expérimentaux de maladie immunitaire de type cellulaire, une réponse humorale dirigée contre les auto-antigènes confère une protection : il a été démontré que les complexes anticorps-antigène dirigent les auto-antigènes vers les cellules B, ce mode de présentation entraînant une réponse cellulaire de type Th2, protectrice [39]. Quand les réponses immunitaires Th1 n’arrivent pas à contrôler la maladie, la maturation des lymphocytes B apporte un changement dans le phénotype de la réponse immunitaire, afin de limiter les dommages inflammatoires, et améliore l’issue du processus immunitaire [40]. Dans notre expérience, les cellules B de souris athéroscléreuses âgées assurent une protection quand elles sont transférées à des souris jeunes, mais susceptibles de développer la maladie (hypercholestérolémiques) [36]. Au contraire, les cellules B de jeunes souris qui n’ont pas encore développé la maladie ne confèrent pas de protection. Les cellules B ne sont donc pas capables de prévenir le déclenchement de la maladie, laquelle est dominée par une réponse immunitaire Th1. Cependant, en parallèle de la progression de la maladie, les souris athéroscléreuses développent une maturation des lymphocytes B qui contribue probablement à réduire la progression des lésions chez les souris âgées.
Conclusions
L’inflammation et la réponse immune participent à la pathogénie de l’athérosclérose dès les stades les plus précoces et peuvent influencer la survenue et l’issue de syndromes coronaires aigus. Les différents composants et phénotypes de la réponse immuno-inflammatoire peuvent jouer un rôle à la fois protecteur et délétère. La variation individuelle dans la réactivité immunitaire, mais aussi différents facteurs environnementaux, peuvent rendre compte de la capacité de certains individus de produire une réponse immunitaire protectrice et efficace quand d’autres ont une réponse immunitaire délétère. La suppression sélective des composants délétères et un renforcement des composants protecteurs du processus inflammatoire lié à l’athérosclérose pourraient fournir de nouveaux outils thérapeutiques pour prévenir et traiter la cause la plus importante de décès dans le monde.

Références
- Braunwald E. Cardiovascular medicine at the turn of the millenium : triumphs, concerns, and opportunities. N Eng J Med 1997; 337 : 1360–9. [Google Scholar]
- Napoli C, D’Armiento FP, Mancini FP, et al. Fatty streak formation occurs in human fetal aortas and is greatly enhanced by maternal hypercholesterolemia. Intimal accumulation of low density lipoprotein and its oxidation precede monocyte recruitment into early atherosclerotic lesions. J Clin Invest 1997; 100 : 2680–90. [Google Scholar]
- Lee RT, Yamamoto C, Feng Y, et al. Mechanical strain induces specific changes in the synthesis and organization of proteoglycans by vascular smooth muscle cells. J Biol Chem 2001; 276 : 13847–51. [Google Scholar]
- Li H, Cybulsky MI, Gimbrone MA Jr, Libby, P. An atherogenic diet rapidly induces VCAM-1, a cytokine-regulatable mononuclear leukocyte adhesion molecule, in rabbit aortic endothelium. Arterioscler Thromb 1993; 13 : 197–204. [Google Scholar]
- Hansson GK. Immune mechanisms in atherosclerosis. Arterioscler Thromb Vasc Biol 2001; 21 : 1876–90. [Google Scholar]
- Virchow R. Phlogose und thrombose in Gefäss-system : Gesammelte Abhandlungen zur Wissenschaftlichen Medicin. Frankfurt-am-Main : Meidinger Sohn, 1856. [Google Scholar]
- Schwartz CJ, Valente AJ, Sprague EA, Kelley JL, Suenram CA, Rozek MM. Atherosclerosis as an inflammatory process. The roles of the monocyte- macrophage. Ann NY Acad Sci 1985; 454 : 115–20. [Google Scholar]
- Kohchi K, Takebayashi S, Hiroki T, Nobuyoshi M. Significance of adventitial inflammation of the coronary artery in patients with unstable angina : results at autopsy Circulation 1985; 71 : 709–16 [Google Scholar]
- Khan BV, Parthasarathy SS, Alexander RW, Medford RM. Modified low density lipoprotein and its constituents augment cytokine-activated vascular cell adhesion molecule-1 gene expression in human vascular endothelial cells. J Clin Invest 1995; 95 : 1262–70. [Google Scholar]
- Cushing SD, Berliner JA, Valente AJ, et al. Minimally modified low density lipoprotein induces monocyte chemotactic protein 1 in human endothelial cells and smooth muscle cells. Proc Natl Acad Sci USA 1990; 87 : 5134–8. [Google Scholar]
- Stemme S, Faber B, Holm J, Wiklund O, Witztum JL, Hansson GK. T lymphocytes from human atherosclerotic plaques recognize oxidized low density lipoprotein. Proc Natl Acad Sci USA 1995; 92 : 3893–7. [Google Scholar]
- Nicoletti A, Caligiuri G, Tornberg I, Kodama T, Stemme S, Hansson GK. The macrophage scavenger receptor type A directs modified proteins to antigen presentation. Eur J Immunol 1999; 29 : 512–21. [Google Scholar]
- Caligiuri G, Nicoletti A, Zhou X, Tornberg I, Hansson GK. Effects of sex and age on atherosclerosis and autoimmunity in apoE- deficient mice. Atherosclerosis 1999; 145 : 301–8. [Google Scholar]
- Farb A, Burke AP, Tang AL, et al. Coronary plaque erosion without rupture into a lipid core. A frequent cause of coronary thrombosis in sudden coronary death. Circulation 1996; 93 : 1354–63. [Google Scholar]
- Davies MJ, Thomas AC. Plaque fissuring - the cause of acute myocardial infarction, sudden ischaemic death, and crescendo angina. Br Heart J 1985; 53 : 363–73. [Google Scholar]
- Friedman GD, Klatsky AL, Siegelaub AB. The leukocyte count as a predictor of myocardial infarction. N Engl J Med 1974; 290 : 1275–8. [Google Scholar]
- De Beer FC, Hind CR, Fox KM, Allan RM, Maseri A, Pepys MB. Measurement of serum C-reactive protein concentration in myocardial ischaemia and infarction. Br Heart J 1982; 47 : 239–43. [Google Scholar]
- Van der Wal AC, Becker AE, van der Loos CM, Das PK. Site of intimal rupture or erosion of thrombosed coronary atherosclerotic plaques is characterized by an inflammatory process irrespective of the dominant plaque morphology. Circulation 1994; 89 : 36–44. [Google Scholar]
- Neri Serneri GG, Abbate R, Gori AM, et al. Transient intermittent lymphocyte activation is responsible for the instability of angina. Circulation 1992; 86 : 790–7. [Google Scholar]
- Caligiuri G, Liuzzo G, Biasucci LM, Maseri A. Immune system activation follows inflammation in unstable angina : pathogenetic implications. J Am Coll Cardiol 1998; 32 : 1295–304. [Google Scholar]
- Liuzzo G, Biasucci LM, Gallimore JR, et al. The prognostic value of C-reactive protein and serum amyloid a protein in severe unstable angina. N Engl J Med 1994; 331 : 417–24. [Google Scholar]
- Caligiuri G, Paulsson G, Nicoletti A, Maseri A, Hansson GK. Evidence for antigen-driven T-cell response in unstable angina. Circulation 2000; 102 : 1114–9. [Google Scholar]
- Itabe H, Takeshima E, Iwasaki H, et al. A monoclonal antibody against oxidized lipoprotein recognizes foam cells in atherosclerotic lesions. Complex formation of oxidized phosphatidylcholines and polypeptides. J Biol Chem 1994; 269 : 15274–9. [Google Scholar]
- Salonen JT, Yla-Herttuala S, Yamamoto R, et al. Autoantibody against oxidised LDL and progression of carotid atherosclerosis. Lancet 1992; 339 : 883–7. [Google Scholar]
- Uyemura K, Demer LL, Castle SC, et al. Cross-regulatory roles of interleukin (IL)-12 and IL-10 in atherosclerosis. J Clin Invest 1996; 97 : 2130–8. [Google Scholar]
- Naruko T, Ueda M, Haze K, et al. Neutrophil infiltration of culprit lesions in acute coronary syndromes. Circulation 2002; 106 : 2894–900. [Google Scholar]
- Biasucci LM, D’Onofrio G, Liuzzo G, et al. Intracellular neutrophil myeloperoxidase is reduced in unstable angina and acute myocardial infarction, but its reduction is not related to ischemia. J Am Coll Cardiol 1996; 27 : 611–6. [Google Scholar]
- Buffon A, Biasucci LM, Liuzzo G, D’Onofrio G, Crea F, Maseri A. Widespread coronary inflammation in unstable angina. N Engl J Med 2002; 347 : 5–12. [Google Scholar]
- Liblau RS, Singer SM, McDevitt HO. Th1 and Th2 CD4+ T cells in the pathogenesis of organ-specific autoimmune diseases. Immunol Today 1995; 16 : 34–8. [Google Scholar]
- Austrup F, Vestweber D, Borges E, et al. P- and E-selectin mediate recruitment of T-helper-1 but not T-helper-2 cells into inflammed tissues. Nature 1997; 385 : 81–3. [Google Scholar]
- Liuzzo G, Goronzy JJ, Yang H, et al. Monoclonal T-cell proliferation and plaque instability in acute coronary syndromes. Circulation 2000; 101 : 2883–8. [Google Scholar]
- Saren P, Welgus HG, Kovanen PT. TNF-alpha and IL-1beta selectively induce expression of 92-kDa gelatinase by human macrophages. J Immunol 1996; 157 : 4159–65. [Google Scholar]
- Caligiuri G, Rudling M, Ollivier V, et al. Interleukin-10 deficiency increases atherosclerosis, thrombosis, and low-density lipoproteins in apolipoprotein E knockout mice. Mol Med 2003; 9 : 10–7. [Google Scholar]
- Laurat E, Poirier B, Tupin E, et al. In vivo downregulation of T helper cell 1 immune responses reduces atherogenesis in apolipoprotein E-knockout mice. Circulation 2001; 104 : 197–202. [Google Scholar]
- Caligiuri G, Stahl D, Kaveri S, et al. Autoreactive antibody repertoire is perturbed in atherosclerotic patients. Lab Invest 2003; 83 : 839–47. [Google Scholar]
- Caligiuri G, Nicoletti A, Poirier B, Hansson GK. Protective immunity against atherosclerosis carried by B cells of hypercholesterolemic mice. J Clin Invest 2002; 109 : 745–53. [Google Scholar]
- Freigang S, Horkko S, Miller E, Witztum JL, Palinski W. Immunization of LDL receptor-deficient mice with homologous malondialdehyde-modified and native LDL reduces progression of atherosclerosis by mechanisms other than induction of high titers of antibodies to oxidative neoepitopes. Arterioscler Thromb Vasc Biol 1998; 18 : 1972–82. [Google Scholar]
- Zhou X, Caligiuri G, Hamsten A, Lefvert AK, Hansson GK. LDL immunization induces T-cell-dependent antibody formation and protection against atherosclerosis. Arterioscler Thromb Vasc Biol 2001; 21 : 108–14. [Google Scholar]
- Saoudi A, Simmonds S, Huitinga I, Mason D. Prevention of experimental allergic encephalomyelitis in rats by targeting autoantigen to B cells : evidence that the protective mechanism depends on changes in the cytokine response and migratory properties of the autoantigen-specific T cells. J Exp Med 1995; 182 : 335–44. [Google Scholar]
- Taylor-Robinson AW, Phillips RS. B cells are required for the switch from Th1- to Th2-regulated immune responses to Plasmodium chabaudi chabaudi infection. Infect Immun 1994; 62 : 2490–8. [Google Scholar]
Liste des tableaux
Rôle possible de marqueur d’inflammation dans l’athérosclérose coronarienne. CRP : protéine C réactive ; SAA : serum amyloid A protein ; CE : cellules endothéliales ; CML : cellules musculaires lisses ; MO : monocytes ; LDL : lipoprotéines de basse densité ; IL : interleukine ; IFN : interféron.
Liste des figures
|
Figure 1. L’hypothèse inflammatoire de l’athérogenèse. Dans l’hypothèse la plus accréditée aujourd’hui, l’extravasation des lipoprotéines de basse densité (LDL) et leur oxydation dans l’espace sous-endothélial représente la première étape de l’athérogenèse. Les LDL oxydées (oxLDL) induisent l’expression de molécules d’adhérence et de récepteurs de chimiokines à la surface des cellules endothéliales (CE), ouvrant ainsi aux monocytes (MO) l’accès à la paroi vasculaire. Une fois entrés, les monocytes-macrophages internalisent les oxLDL, les accumulent à l’intérieur et se transforment ainsi en cellules spumeuses. Des lymphocytes T (LT) sont également recrutés, probablement alertés par les cytokines sécrétées par les macrophages (MP) et par des molécules d’adhérence et des chimiokines exprimées par les cellules endothéliales et musculaires lisses (CML) activées. Ces lymphocytes sécrètent des cytokines et expriment des marqueurs d’activation, qui signent vraisemblablement une stimulation antigénique locale, pouvant entraîner des effets importants sur le devenir de plaques précoces, ainsi que sur la biologie de la paroi artérielle et la pathogénie des complications des plaques (d’après [5]). |
| Dans le texte | |
|
Figure 2. ’hypothèse inflammatoire dans les syndromes coronariens aigus. Une réponse immuno-inflammatoire aiguë et transitoire est détectable dans le sang circulant des patients présentant des syndromes coronariens aigus. L’activation des lymphocytes T et B (production d’anticorps, Ac) suggère que l’instabilité pourrait avoir été déclenchée par des stimulus antigéniques (comme l’indique la présence de pics monoclonaux dans l’analyse de l’ARNm codant pour le récepteur de l’antigène des lymphocytes T activés, par la technique de l’immunoscope), et que la réponse immuno-inflammatoire associée pourrait participer directement à la pathogénie de l’instabilité. Cette réponse immuno-inflammatoire devient détectable peu de temps après la survenue des symptômes dans l’angor instable. En fait, certains antigènes cibles contenus dans les plaques ne deviendraient accessibles aux lymphocytes circulants que lors d’une complication telle que la rupture de la chape fibreuse. Cela pourrait indiquer que la réponse immuno-inflammatoire observée dans les phases d’instabilité de l’athérosclérose coronaire pourrait être une conséquence, plutôt qu’une cause, de l’instabilité clinique. Dans ce cas, il reste plausible que la réponse immuno-inflammatoire joue un rôle important dans l’issue de l’instabilité clinique. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.