")
")
| Issue |
Med Sci (Paris)
Volume 19, Number 10, Octobre 2003
|
|
|---|---|---|
| Page(s) | 926 - 930 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/20031910926 | |
| Published online | 15 October 2003 | |
Expression du gène 17β-HSD2 dans le poumon fœtal et le placenta : contrôle de l’action des stéroïdes sexuels
Expression of HSD17β2 gene in the fetal lung and placenta: control of sex steroid action
Unité d’Ontogénie et Reproduction, Centre hospitalier universitaire Laval (CHUL), Département d’Obstétrique et Gynécologie, Faculté de Médecine, Université Laval, 2705, boulevard Laurier, Sainte-Foy, Québec, G1V 4G2,Canada
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
Durant la grossesse, les stéroïdes sexuels pourraient potentiellement nuire au développement fœtal. Deux mécanismes distincts au moins, faisant intervenir la 17β-hydroxystéroïde déshydrogénase- 2 (17β-HSD2), permettraient d’empêcher ces actions indésirables. En s’exprimant dans les fibroblastes pulmonaires et les cellules endothéliales placentaires, la 17β-HSD2 limiterait respectivement l’action de la testostérone et de l’œstradiol, permettant ainsi un développement normal.
Abstract
During gestation, sex steroids could potentially have detrimental effects on fetal development. At least two distinct mechanisms should prevent such effects. The 17β-hydroxysteroid dehydrogenase-2 (HSD17β2) plays a key role in each mechanism. Being expressed both in lung fibroblasts and placental endothelial cells, the HSD17β2 should restrict testosterone and estradiol actions, thus enabling normal development.
© 2003 médecine/sciences - Inserm / SRMS
Chez l’homme, les surrénales sécrètent des stéroïdes précurseurs qui seront transformés en stéroïdes sexuels actifs par les tissus cibles extra-gonadiques [1]. Cette découverte majeure a donné une nouvelle perspective à l’étude des mécanismes d’action des hormones stéroïdiennes en introduisant le concept d’une action intra/autocrine et paracrine des stéroïdes. En effet, les stéroïdes produits par ces tissus transmettront, via leurs récepteurs spécifiques, un signal directement à la cellule productrice dans le cas d’une action intra/autocrine, ou à une cellule voisine dans le cas d’une action paracrine. La présence des androgènes et des oestrogènes comme facteurs paracrines est certes bénéfique, mais peut, dans certains cas, être dommageable. Dans le poumon, la testostérone serait produite par les cellules alvéolaires (pneumocytes de type II, PTII) productrices de surfactant comme cela est démontré par nos études sur les cellules A549 [2]. De plus, il est clairement rapporté dans la littérature que les androgènes présents dans le poumon retardent le développement pulmonaire et la production de surfactant par les cellules PTII [3]. Ainsi, une fréquence plus élevée de détresses respiratoires est enregistrée chez les garçons nés avant terme que chez les filles, le développement pulmonaire des garçons étant retardé par la présence d’une concentration plus élevée d’androgènes dans le poumon (pour revue, voir [3]). Par ailleurs, il est bien connu que les oestrogènes peuvent avoir des effets indésirables à certains moments du développement fœtal [4], comme par exemple, des anomalies anatomiques dans le développement du tractus génital mâle et femelle. En outre, les filles dont les mères avaient été exposées aux oestrogènes durant la grossesse ont également plus de risque d’avoir une grossesse à problèmes. En effet, une étude conduite par S.H. Swan en 1992 concluait que les filles préalablement exposées aux oestrogènes étaient plus à risque de grossesses ectopiques, d’accouchements prématurés et d’avortements que les femmes qui n’y ont jamais été exposés [5]. Cependant, un développement normal du foetus est observé même en présence d’une production massive d’oestradiol par les trophoblastes placentaires [6, 7]. Ces deux observations, en apparence contradictoires, pourraient s’expliquer par la présence d’un mécanisme visant à contrôler la concentration d’oestrogènes dans la circulation fœtale. En fait, dans le cas d’une grossesse normale, les effets délétères des androgènes sur le développement pulmonaire et des estrogènes sur le développement fœtal seraient limités par deux mécanismes de contrôle faisant intervenir le même gène, celui de la 17β- hydroxystéroïde déshydrogénase-2 (17β-HSD2) [8, 9]. En effet, la 17β-HSD2, possédant des activités d’inactivation des androgènes (conversion de la testostérone en androstènedione) et des oestrogènes (conversion d’oestradiol en œstrone), est présente dans le poumon fœtal et le placenta. Dans le poumon, la 17β-HSD2 est exprimée dans les fibroblastes, une population cellulaire jouant un rôle clé dans la maturation pulmonaire [9]. L’inactivation des androgènes par la 17β-HSD2 favoriserait la maturation pulmonaire. Dans le placenta, l’expression de la 17β-HSD2 est spécifique aux cellules endothéliales qui forment la paroi des vaisseaux sanguins situés dans les villosités placentaires. L’oestradiol doit obligatoirement traverser ces cellules endothéliales pour accéder à la circulation fœtale [8, 10]. En conséquence, le niveau d’oestradiol sécrété dans la circulation fœtale doit être contrôlé par la 17β-HSD2 qui est exprimée dans les EC.
Synthèse et inactivation des androgêns par le poumon fœtal
La proportion de naissances prématurées est de 10% et 75% de celles-ci sont associées à une morbidité néonatale [11, 12]. Parmi les complications possibles, la détresse respiratoire préoccupe beaucoup le néonatalogiste. Ce syndrome survient lorsque la naissance se produit avant la maturation complète des cellules PTII et est favorisé par les androgènes qui retardent la maturation pulmonaire [3]. Les garçons représentent 70% des cas de détresse respiratoire et sont exposés à des concentrations plus élevées d’androgènes pulmonaires. La maturation des cellules PTII conduisant à la production de surfactant s’effectue dans le dernier tiers de la grossesse et est stimulée par des facteurs paracrines sécrétés par les fibroblastes [13, 14]. Des études in vitro ont montré que les androgènes inhibaient, via le récepteur des androgènes [15], la sécrétion par les fibroblastes de ces facteurs paracrines essentiels à la maturation des cellules PTII. Nous avons montré que les cellules A-549, une lignée épithéliale productrice de surfactant et issue de cellules PTII d’origine mâle, ont une très forte activité de conversion de l’androstènedione en testostérone catalysée par la 17β-HSD5. Ces cellules sécrètent de la testostérone qui s’accumule en raison d’une faible activité 5α-réductase [2]. Ces expériences suggèrent que les cellules PTII produiraient et sécréteraient de la testostérone in vivo comme androgène actif. C’est pourquoi nous avons également étudié la capacité des fibroblastes pulmonaires à inactiver la testostérone. Aucune activité de synthèse d’androgènes n’a été détectée dans huit lignées primaires de fibroblastes humains d’origine mâle ou femelle, isolées durant le second trimestre de gestation ou durant la période néonatale. Cependant, toutes les lignées avaient la propriété commune d’inactiver la testostérone en exprimant la 17β- HSD2 et de transformer le produit de cette réaction (l’androstènedione) via la 5α-réductase-1 et ce, sans former de 5α-dihydrotestostérone, l’androgène le plus puissant, généralement produit par la 5α-réductase à partir de la testostérone. Ces lignées ont été classées en deux groupes selon le niveau d’activité de la 17β-HSD2. Certaines lignées ont en effet une activité très faible comparativement aux autres. Une étude sur quelques échantillons de poumon humain adulte a également montré une expression similaire de la 17β-HSD2 avec des niveaux variables d’ARNm d’un sujet à l’autre [9]. Nous avons donc proposé l’existence d’une variation allélique affectant le niveau d’expression de la 17β-HSD2. Ces données prennent toute leur importance si l’on considère que les androgènes retardent la maturation pulmonaire par une action sur les fibroblastes et que le métabolisme stéroïdien des fibroblastes est orienté exclusivement vers une inactivation des androgènes. Ainsi, la 17β-HSD2 favoriserait la maturation pulmonaire en inactivant la testostérone spécifiquement dans les cellules où les androgènes pourraient inhiber le mécanisme de maturation (Figure 1). En contrepartie, les sujets présentant un faible niveau de 17β-HSD2 pourraient être prédisposés au risque de faire une détresse respiratoire. En résumé, l’androstènedione d’origine surrénalienne serait transformée en testostérone par les cellules PTII. La testostérone serait sécrétée dans l’environnement cellulaire où, par action paracrine, elle pourrait inhiber la sécrétion par les fibroblastes de certains facteurs impliqués dans la maturation des cellules PTII. Cependant, l’expression de la 17β-HSD2 à un niveau suffisant dans les fibroblastes permettrait d’empêcher cet effet des androgènes.
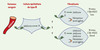 |
Figure 1. Schéma présentant le modèle de la stéroïdogenèse dans les cellules épithéliales alvéolaires PTII et dans les fibroblastes pulmonaires. Les surrénales sécrètent des stéroïdes précurseurs qui, via la circulation sanguine, deviennent des substrats pour les tissus cibles. Dans ce modèle, l’androstènedione (∆4dione) est transformé en testostérone (T) par les cellules PTII. La testostérone produite est sécrétée dans le stroma interstitiel et constitue un substrat pour les fibroblastes. Selon notre hypothèse, les fibroblastes seraient divisés en deux catégories: ceux ayant des niveaux élevés et ceux ayant des niveaux bas d’activité 17β-HSD type 2. Le niveau d’activité 17 β -HSD type 2 dans les fibroblastes déterminerait les concentrations d’androgènes accessibles aux récepteurs des androgènes. |
Inactivation des oestrogènes par le placenta humain
Des publications concernant plusieurs milliers de femmes enceintes, auxquelles des oestrogènes avaient été administrés, rapportent qu’une exposition à des quantités excessives d’estrogènes a des conséquences néfastes chez le foetus en développement et plus tard à l’âge adulte [4, 5]. Cependant, durant la grossesse normale, l’oestradiol est produit par le placenta en quantité nettement suffisante pour lier le récepteur des oestrogènes. Ces observations suggèrent fortement l’existence d’un mécanisme protégeant le foetus des conséquences d’une exposition aux quantités excessives d’oestrogènes produites dans le placenta. Par ailleurs, tardivement durant la gestation, mais à des périodes semblables de développement fœtal, l’oestradiol est nécessaire au développement de certains systèmes comme la peau [16] et les poumons [17]. Ceci suggère que le besoin du foetus en oestradiol varie selon le stade de développement fœtal. Ces observations a priori contradictoires pourraient s’expliquer par l’existence d’un mécanisme contrôlant la concentration d’oestradiol dans la circulation fœtale. Nous avons montré que la 17β-HSD2, une déshydrogénase spécialisée dans l’inactivation des stéroïdes sexuels, pourrait jouer ce rôle. L’oestradiol est produit presque exclusivement par le syncytiotrophoblaste [6, 18, 19]. Le syncytium forme la couche cellulaire la plus externe de la villosité choriale. À l’intérieur de la villosité, se trouvent des vaisseaux sanguins (veines et artères) qui sont le prolongement du système circulatoire fœtal. Il est à noter que le sang oxygéné parvient au foetus par le système veineux. Tout stéroïde doit obligatoirement franchir la paroi de ces vaisseaux pour accéder à la circulation fœtale. Donc, l’expression de la 17β-HSD2 dans les parois de ces vaisseaux serait un mécanisme potentiel de protection du foetus contre des quantités excessives d’oestradiol. Deux études indépendantes [10, 20] utilisant un anticorps anti-17β-HSD2 ont rapporté la présence de cette enzyme dans près de 100% des cellules endothéliales de placentas à mi-gestation. Par hybridation in situ, nous avons montré que le gène de la 17β-HSD2 était exprimé exclusivement par les cellules endothéliales dans le placenta à terme [8]. De plus, nous avons clairement établi que la très grande majorité des veines n’exprimaient pas la 17β-HSD2 à terme. Sachant que les étapes de développement qui requièrent la présence d’œstradiol se déroulent durant le dernier tiers de la gestation, nos résultats suggèrent l’existence d’un mécanisme (Figure 2) par lequel les quantités d’oestradiol parvenant au foetus via la circulation veineuse seraient modulées tout au long de la gestation de façon à permettre le passage d’une grande quantité d’oestradiol vers la fin de la grossesse, et en limitant à un stade plus précoce le passage de cette hormone, prévenant ainsi son effet néfaste sur le développement.
 |
Figure 2. Schéma présentant les activités enzymatiques des isoformes 17β-HSD type 1 et 17 β -HSD type 2 en relation avec le contrôle par le placenta des concentrations d’oestradiol dans la circulation fœtale. La 17 β -HSD type 1 est une enzyme soluble présente dans les syncytiotrophoblastes et dont l’activité prédominante est de nature réductase avec une spécificité pour les oestrogènes (conversion de l’oestrone en oestradiol). En revanche, la 17 β -HSD type 2 est une enzyme membranaire présente dans le réticulum endoplasmique et dont l’activité déshydrogénase présente des affinités semblables pour les oestrogènes et les androgènes. Dans le placenta, la 17β-HSD type 2 a été exclusivement détectée dans les cellules endothéliales formant les vaisseaux sanguins des villosités. Nos résultats suggèrent l’existence d’un mécanisme par lequel les quantités d’oestradiol parvenant au foetus via la circulation veineuse seraient modulées tout au long de la gestation de façon à permettre le passage d’une grande quantité d’oestradiol vers la fin de la grossesse et en limitant à un stade plus précoce le passage de cette hormone, empêchant ainsi son effet néfaste sur le développement. E2: oestradiol; 17 β -1: 17 β -HSD type 1; E1: oestrone;17 β -2 : 17 β -HSD type 2. |
Conclusions
Dans ces deux modèles, l’action des stéroïdes sexuels serait modulée par la 17β-HSD2 exprimée respectivement par deux types cellulaires différents. Ainsi, le poumon fœtal et le placenta auraient chacun un mécanisme distinct de contrôle de l’action des stéroïdes sexuels faisant intervenir le même gène. La présence de la 17β-HSD2 dans ces deux tissus semble jouer un rôle identique : permettre un développement normal en contrôlant l’exposition de certaines cellules ou organes aux stéroïdes sexuels.
Remerciements
Nous remercions Martin Bonenfant, Renée Drolet et Marc Simard, étudiants diplômés du laboratoire, ainsi que le Dr Charles H. Blomquist.
Références
- Labrie F, Bélanger A, Dupont A, et al. Synthèse périphérique des androgènes chez l’homme. Med Sci 1990; 6: 261–7. [Google Scholar]
- Provost PR, Blomquist CH, Godin C, et al. Androgen formation and metabolism in the pulmonary epithelial cell line A-549: expression of 17β-hydroxysteroid dehydrogenase type 5 and 3α-hydroxysteroid dehydrogenase type 3. Endocrinology 2000; 141: 2786–94. [Google Scholar]
- Ballard PL. Hormonal regulation of pulmonary surfactant. Endocrinol Rev 1989; 10: 165–81. [Google Scholar]
- Giusti RM, Iwamoto K, Hatch EE. Diethylstilbestrol revisited: a review of the long-term health effects. Ann Intern Med 1995; 122: 778–88. [Google Scholar]
- Swan SH. Pregnancy outcome in DES daughters. In: Giusti RM, ed. Report of the NIH Workshop on longterm effects of exposure to diethylstilbestrol (DES). Washington, DC: US Department of Health and Human Services, Public Health Service, National Institutes of Health, 1992: 420–49. [Google Scholar]
- Bonenfant M, Provost PR, Drolet R, Tremblay Y. Localization of type 1 17β- hydroxysteroid dehydrogenase mRNA and protein in syncytiotrophoblasts and invasive cytotrophoblasts in the human term villi. J Endocrinol 2000; 165: 217–22. [Google Scholar]
- Beaudoin C, Blomquist CH, Bonenfant M, Tremblay Y. Expression of the genes for 3β-hydroxysteroid dehydrogenase type 1 and cytochrome P450scc during syncytium formation hy human placental cytotrophoblast cells in culture and its regulation by progesterone and estradiol. J Endocrinol 1997; 154: 379–87. [Google Scholar]
- Bonenfant M, Blomquist CH, Provost PR, Drolet R, D’Ascoli P, Tremblay Y. Tissue- and site-specific expression of type 2 17β- hydroxysteroid dehydrogenase: in situ hybridization and specific enzymatic activity studies in human placental endothelial cells of the arterial system. J Clin Endocrinol Metab 2000; 85: 4841–50. [Google Scholar]
- Provost PR, Blomquist CH, Drolet R, Flamand N, Tremblay Y. Androgen inactivation in human lung fibroblasts: variations in levels of 17β- hydroxysteroid dehydrogenase type 2 and 5α-reductase activity compatible with androgen inactivation. J Clin Endocrinol Metab 2002; 87: 3883–92. [Google Scholar]
- Takeyama J, Sasano H, Suzuki T, Iinuma K, Nagura H, Andresson S. 17β- hydroxysteroid dehydrogenase types 1 and 2 in human placenta; an immunohistochemical study with correlation to placental development. J Clin Endocrinol Metab 1998; 83: 3710–5. [Google Scholar]
- Farrell PM, Avery ME. Hyaline membrane disease. Am Rev Respir Dis 1975; 111: 657–88. [Google Scholar]
- Miller HC, Futrakul P. Birthweight, gestational age, and sex as determining factors in the incidence of respiratory distress syndrome of prematurely born infants. Pediatrics 1968; 72: 628–35. [Google Scholar]
- Post M, Barsoumian A, Smith BT. The cellular mechanism of glucocorticoid acceleration of fetal lung maturation. Fibroblast-pneumonocyte factor stimulates cholinephosphate cytidylyltransferase activity. J Biol Chem 1986; 261: 2179–84. [Google Scholar]
- Minkowski A, Monset- Couchard M. The physiological and biochemical basis for perinatal medicine. New York: Karger, 1981: 370 p. [Google Scholar]
- Nielsen HC, Zinman HM, Torday JS. Dihydrotestosterone inhibits fetal rabbit pulmonary surfactant production. J Clin Invest 1982; 69: 611–6. [Google Scholar]
- Hanley K, Rassner U, Jiang Y, et al. Hormonal basis for the gender difference in epidermal barrier formation in the fetal rat. Acceleration by estrogen and delay by androgen. J Clin Invest 1996; 97: 2576–84. [Google Scholar]
- Adamson IY, Bakowska J, McMillan E, King GM. Accelerated fetal lung maturation by estrogen is associated with an epithelial-fibroblast interaction. In Vitro Cell Dev Biol 1990; 26: 784–90. [Google Scholar]
- Beaudoin C, Blomquist CH, Tremblay Y. Gene expression of 17β- hydroxysteroid dehydrogenase type 2 isozyme in primary cultures of human trophoblasts predicts different mechanisms regulating type 1 and type 2 enzymes. Endocrinology 1995; 136: 3807–14. [Google Scholar]
- Simpson ER, Mahendroo MS, Means GD, et al. Aromatase cytochrome P450, the enzyme responsible for estrogen biosynthesis. Endocrinol Rev 1994; 15: 342–55. [Google Scholar]
- Moghrabi N, Head JR, Andersson S. Cell type specific expression of human 17β-hydroxysteroid dehydrogenase type 2 in human placenta and fetal liver. J Clin Endocrinol Metab 1997; 82: 3872–8. [Google Scholar]
Liste des figures
|
Figure 1. Schéma présentant le modèle de la stéroïdogenèse dans les cellules épithéliales alvéolaires PTII et dans les fibroblastes pulmonaires. Les surrénales sécrètent des stéroïdes précurseurs qui, via la circulation sanguine, deviennent des substrats pour les tissus cibles. Dans ce modèle, l’androstènedione (∆4dione) est transformé en testostérone (T) par les cellules PTII. La testostérone produite est sécrétée dans le stroma interstitiel et constitue un substrat pour les fibroblastes. Selon notre hypothèse, les fibroblastes seraient divisés en deux catégories: ceux ayant des niveaux élevés et ceux ayant des niveaux bas d’activité 17β-HSD type 2. Le niveau d’activité 17 β -HSD type 2 dans les fibroblastes déterminerait les concentrations d’androgènes accessibles aux récepteurs des androgènes. |
| Dans le texte | |
|
Figure 2. Schéma présentant les activités enzymatiques des isoformes 17β-HSD type 1 et 17 β -HSD type 2 en relation avec le contrôle par le placenta des concentrations d’oestradiol dans la circulation fœtale. La 17 β -HSD type 1 est une enzyme soluble présente dans les syncytiotrophoblastes et dont l’activité prédominante est de nature réductase avec une spécificité pour les oestrogènes (conversion de l’oestrone en oestradiol). En revanche, la 17 β -HSD type 2 est une enzyme membranaire présente dans le réticulum endoplasmique et dont l’activité déshydrogénase présente des affinités semblables pour les oestrogènes et les androgènes. Dans le placenta, la 17β-HSD type 2 a été exclusivement détectée dans les cellules endothéliales formant les vaisseaux sanguins des villosités. Nos résultats suggèrent l’existence d’un mécanisme par lequel les quantités d’oestradiol parvenant au foetus via la circulation veineuse seraient modulées tout au long de la gestation de façon à permettre le passage d’une grande quantité d’oestradiol vers la fin de la grossesse et en limitant à un stade plus précoce le passage de cette hormone, empêchant ainsi son effet néfaste sur le développement. E2: oestradiol; 17 β -1: 17 β -HSD type 1; E1: oestrone;17 β -2 : 17 β -HSD type 2. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.