")
")
| Issue |
Med Sci (Paris)
Volume 19, Number 8-9, Août-Septembre 2003
|
|
|---|---|---|
| Page(s) | 854 - 859 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/20031989854 | |
| Published online | 15 août 2003 | |
Le MODY : modèle d’étude d’interactions génotype/phénotype dans le diabète de type 2
MODY, a model of genotype/phenotype interactions in type 2 diabetes
1
Inserm U.561 82, avenue Denfert-Rochereau, 75014 Paris, France
2
Laboratoire d’embryologie pathologique-cytogénétique, Unité de génétique, Hôpital Saint-Antoine, 184, rue du Faubourg Saint-Antoine, 75571 Paris Cedex 12, France
3
Unité de diabétologie, Service d’immunologie clinique, Hôpital Necker-Enfants Malades, 149, rue de Sèvres, 75743 Paris Cedex 15, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Le MODY (maturity onset diabetes of the young) est une forme de diabète familial, à transmission autosomique dominante et à début précoce, associé à des anomalies primaires de l’insulinosécrétion. Des mutations dans six gènes (l’enzyme glucokinase et cinq facteurs de transcription exprimés dans le pancréas) sont responsables de la plupart des cas de MODY. Cette hétérogénéité génétique est associée à une hétérogénéité métabolique et clinique faisant du MODY un modèle intéressant d’étude d’interactions génotype/ phénotype dans le diabète.
Abstract
Maturity onset diabetes of the young (MODY) is a subtype of familial diabetes mellitus characterised by early onset, autosomal dominant inheritance and primary defects of insulin secretion. Mutations in six known genes (the enzyme glucokinase and five transcription factors expressed in pancreatic β-cells) cause most of the MODY cases. This genetic heterogeneity is associated with metabolic and clinical heterogeneity making MODY an interesting model of genotype/phenotype interaction in diabetes.
© 2003 médecine/sciences - Inserm / SRMS
Le diabète de type 2 est une maladie multifactorielle complexe, où facteurs génétiques polygéniques et facteurs d’environnement sont étroitement associés. Il existe cependant de nombreux cas dans lesquels la mutation d’un seul gène est suffisante pour conduire à une hyperglycémie. Le diabète de type MODY (maturity onset diabetes of the young), le plus fréquent des diabètes monogéniques, est défini sur le phénotype suivant [1, 2]: diabète familial présentant une transmission autosomique dominante, hyperglycémie de survenue précoce (classiquement avant l’âge de 25 ans, mais souvent à l’enfance ou à l’adolescence) cliniquement non insulinodépendante, au moins pendant les premières années suivant le diagnostic, et présence d’une anomalie primaire de l’insulinosécrétion (→).
(→) m/s 1998, n°3, p. 364
Bien que monogénique, le MODY est génétiquement hétérogène : des mutations hétérozygotes dans six gènes ont à ce jour été identifiées à l’origine d’un MODY (Tableau I). Cinq de ces gènes codent pour des facteurs de transcription (MODY-1, 3, 4, 5 et 6) exprimés dans les cellules β-pancréatiques. Le MODY-2 est, quant à lui, dû à des mutations du gène de l’enzyme glucokinase. Tous types confondus, le MODY représenterait 2 à 5 % des diabètes non insulinodépendants, avec une très forte prédominance des MODY-2 et 3, les autres formes ne concernant jusqu’ici que quelques familles. Tous les gènes du MODY ne sont pas actuellement identifiés, permettant d’envisager un démembrement ultérieur du sous-groupe MODY-X.
Sous-types de MODY. * Distribution selon les populations ; Tm : taux maximal de réabsorption ; HNF : hepatocyte nuclear factor; IPF-1 : insulin promoter factor 1 ; Neuro-D1/β2 : neurogenic differentiation factor 1/β-cell E-box transactivator 2 ; Glut2 : glucose transporter 2 ; L-PK : liver-specific pyruvate kinase ; AldoB : aldolase B ; 1,3-BGD : glycéraldéhyde-3-phosphate déshydrogénase ; SGLT2 : sodium glucose transporter 2 ; NBAT : neutral and basic amino acid transporter ; PCD/DCoH : ptérine-4α-carbinolamine déshydratase ; IAPP : islet amyloid polypeptide.
Ce cadre nosologique est en réalité très hétérogène en ce qui concerne les anomalies moléculaires, le degré d’altération de l’insulinosécrétion et son caractère évolutif ou non, la sévérité de l’hyperglycémie qui en résulte et donc la fréquence des complications associées [1, 2]. Même si la physiopathologie des MODY reste incomplètement élucidée, le démembrement moléculaire de ces formes de diabète a permis d’établir le lien entre les anomalies génétiques, les défauts métaboliques et la présentation clinique de ces formes de diabète. Dans ce contexte d’hétérogénéité génétique, métabolique et clinique, le MODY est un modèle intéressant d’étude d’interactions génotype/phénotype dans le diabète de type 2.
Le diabéte MODY-2 par déficience en glucokinase
La glucokinase catalyse la phosphorylation du glucose en glucose-6-phosphate dans les cellules insulinosécrétoires du pancréas endocrine et dans les hépatocytes: elle constitue dans ces tissus une enzyme clé du contrôle du métabolisme du glucose (Figure 1). Les mutations de la glucokinase associées au diabète diminuent l’activité enzymatique de phosphorylation du glucose. Or, dans les cellules β-pancréatiques, le métabolisme du glucose et l’insulinosécrétion sont fortement dépendants de l’activité de l’enzyme. On observe, chez les sujets porteurs des mutations, une diminution de moitié environ de la sensibilité des cellules β-pancréatiques au glucose. Cela se traduit par une élévation du seuil glycémique induisant la libération de l’insuline, par un décalage vers la droite de la courbe dose/réponse de la sécrétion d’insuline en fonction de la glycémie et par une diminution de moitié de la quantité d’insuline libérée pour un niveau glycémique donné [3, 4]. Dans le foie, la glucokinase joue un rôle important dans le captage hépatique postprandial et le stockage du glucose sous forme de glycogène (Figure1). La synthèse postprandiale du glycogène hépatique est diminuée, et la production hépatique de glucose augmentée chez les porteurs de mutations de la glucokinase [5].
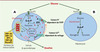 |
Figure 1. Représentation schématique du rôle régulateur des gènes du MODY dans l’homéostasie glycémique. A. Implication dans la sécrétion d’insuline en réponse au glucose: (a) phosphorylation du glucose en G-6-P par la glucokinase; (b) production d’ATP par la glycolyse et par le cycle de Krebs; (c) augmentation du rapport ATP/ADP intracellulaire ; (d) fermeture des canaux potassiques ATP-dépendants; (e) augmentation de la concentration intracellulaire de K+; (f) dépolarisation de la membrane cellulaire; (g) ouverture des canaux calciques dépendants du voltage ; (h) augmentation de la concentration intracellulaire de Ca2+ ; (i) exocytose de l’insuline. B. Implication de la glucokinase dans la synthèse postprandiale de glycogène hépatique. |
Les mutations du gène de la glucokinase représentent la cause la plus fréquente de MODY en France (50 % des cas). Les anomalies métaboliques du MODY-2 sont présentes dès la naissance et même probablement in utero. En effet, dans des familles MODY-2, le poids de naissance des enfants porteurs d’une mutation est réduit par rapport à celui des nouveau-nés non porteurs de la mutation, ce qui suggère une réduction de l’insulinosécrétion et donc de la croissance in utero [6]. Cette hypothèse est confortée par les données obtenues dans un modèle de souris dont un allèle du gène codant pour la glucokinase a été invalidé. La pénétrance du MODY-2 est complète : dans une famille, tous les sujets porteurs de la mutation sont hyperglycémiques [7]. Dans la majorité des cas, l’hyperglycémie associée aux mutations de la glucokinase est modérée et très stable dans le temps [7, 8]. En conséquence, les complications sévères de microangiopathie sont peu fréquentes dans cette forme de diabète, moins que dans le diabète de type 2 classique ou dans d’autres formes de MODY : moins de 5 % des patients développent une rétinopathie proliférative, une néphropathie clinique ou une neuropathie périphérique [7, 9]. De même, ces patients n’ont habituellement pas les facteurs de risque vasculaire habituellement associés au diabète de type 2 et les complications de macroangiopathie sont rares. Ces données soulignent l’intérêt du diagnostic moléculaire de MODY-2 qui permet de préciser le pronostic du diabète chez les patients et de guider le dépistage familial.
Le diabéte MODY-3 dû aux mutations d’HNF-1α (hepatocyte nuclear factor 1-α)
Le diabète de type MODY-3 se distingue radicalement du MODY-2. Des anomalies sévères de l’insulinosécrétion sont observées chez les patients porteurs de mutation d’HNF-1α (hepatocyte nuclear factor 1-α) [10] et l’hyperglycémie est beaucoup plus marquée que celle observée au cours du MODY2 [8]. La sensibilité à l’insuline est altérée chez ces patients diabétiques, secondairement à la carence en insuline et à l’hyperglycémie. La physiopathologie du MODY-3 reste incomplètement élucidée. Le gène codant pour HNF-1α est exprimé dans divers tissus, dont le foie, le rein, le pancréas et le tube digestif. Plus d’une centaine de mutations du gène HNF-1α ont été identifiées dans des familles MODY-3, siégeant dans les domaines fonctionnels du gène ou dans le promoteur [1]. L’étude des souris dont le gène codant pour HNF-1α a été invalidé a fourni des informations sur les mécanismes de survenue du diabète [11, 12]. Chez ces souris, il existe une hyperglycémie en rapport avec une diminution sévère (de 85 %) de la réponse insulinosécrétoire au glucose. La taille du pancréas et la masse de cellules β semblent en rapport avec le poids très réduit des animaux. Globalement, l’expression de nombreux gènes impliqués dans le développement des îlots et dans leur métabolisme, notamment dans la production mitochondriale d’ATP, est altérée (Tableau I). Les études fonctionnelles, dans des systèmes in vitro, de certains des mutants identifiés ont montré qu’une diminution de la liaison à l’ADN ou une perte de l’activité de transactivation peuvent être en cause dans les anomalies de l’insulinosécrétion [13].
L’expression phénotypique du MODY-3 est très variable d’une famille à l’autre et au sein d’une même famille. Par opposition au MODY-2 dont la pénétrance est complète, certains sujets porteurs d’une mutation du gène HNF-1α sont normoglycémiques, alors que leurs germains d’âge comparable sont très hyperglycémiques. Ces porteurs «sains» de la mutation présentent néanmoins des anomalies de l’insulinosécrétion [10] et peuvent développer ultérieurement un diabète, en particulier dans des situations d’insulinorésistance, comme la grossesse ou une prise de poids. Le début du diabète survient, dans la majorité des cas, dans la période post-pubertaire (âge moyen au diagnostic: 22 à 26 ans selon les séries), contrairement au MODY-2. La plupart des sujets présentent un syndrome polyuro-polydipsique franc lors du diagnostic. Il existe en effet une diminution de 50 % environ du taux maximal de réabsorption (Tm) rénale du glucose qui majore ce syndrome (et limite peut-être l’hyperglycémie) [14]. La diminution d’activité d’HNF-1α altère l’expression du transporteur sodium-glucose SGLT2 du tubule proximal. Cette anomalie est présente chez les sujets encore normoglycémiques porteurs de mutations d’HNF-1α et pourrait rendre compte de l’observation ancienne de glucosurie précédant la survenue d’un diabète. Les complications de microangiopathie sont fréquentes au cours du MODY-3. À durée d’évolution et sévérité de l’hyperglycémie comparables, la prévalence de ces complications est la même qu’au cours des diabètes de type 1 et de type 2 [9, 15]. En revanche, la prévalence de l’hypertension artérielle, de l’obésité, des anomalies lipidiques et des complications de macroangiopathie (maladie coronarienne) est plus faible qu’au cours du diabète de type 2 [15].
Les autres formes de MODY
Le MODY-1 est dû aux mutations du gène codant pour HNF-4α, un membre orphelin de la superfamille des récepteurs nucléaires. Le MODY-1 semble rare (une quinzaine de familles décrites à ce jour), mais sa description ancienne et le suivi prospectif de la très grande famille connue sous le nom de RW a fourni des informations intéressantes [1]. Dans cette famille, le diabète apparaît à un âge variable, entre 7 et 40 ans, mais une hyperglycémie survient à terme chez 95% des sujets porteurs de la mutation. L’évolution se fait vers une perte progressive de l’insulinosécrétion, dont témoigne une réponse aux sulfamides hypoglycémiants qui diminue de 1 à 4 % par an. Environ 40 % des patients ne répondent plus aux sulfamides hypoglycémiants après 3 à 25 ans d’évolution, et deviennent «instables » comme des diabétiques de type 1. À l’inverse, certains sujets ont pu être bien contrôlés par ce traitement pendant 40 ans.
Les mécanismes moléculaires par lesquels une réduction d’activité HNF-4α aboutit à un défaut d’insulinosécrétion et au diabète ne sont pas clairement déterminés. Une étude dans des cellules souches d’origine embryonnaires a démontré que la perte de fonction d’HNF-4α est associé à une diminution de l’expression de nombreux gènes impliqués dans le métabolisme du glucose [16] (Tableau I). Les mécanismes physiopathologiques des anomalies de l’insulinosécrétion au cours du MODY-1 sont probablement proches de ceux qui sont impliqués dans le MODY-3, car HNF-4α est l’un des facteurs réglant la transcription d’HNF-1α. Réciproquement, il a été récemment rapporté l’existence d’un second promoteur d’HNF-4α comportant des sites de liaison pour HNF-1α, HNF- 1β et IPF-1 [17]. Une mutation du site de liaison d’IPF-1 co-ségrégeant avec le diabète a été décrite. Ces résultats suggèrent l’existence d’un réseau interconnectant divers facteurs de transcription impliqués dans le développement et la fonction des îlots pancréatiques (Figure 2) [16–18]. Ils suggèrent également la présence de mécanismes physiopathologiques communs dans les différentes formes de MODY.
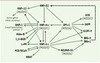 |
Figure 2. Représentation schématique du réseau interconnectant les facteurs de transcription impliqués dans le MODY et leurs gènes cibles. Les flèches représentent des régulations transcriptionelles positives ou négatives dans le cas de l’effet de SHP-1 sur HNF4α. HNF-1α et HNF-1β forment des homo- et des hétérodimères. HNF: hepatocyte nuclear factor ; SHP-1: small heterodimer partner 1; AldoB: aldolase B; LPK: liver-specific-pyruvate kinase; PCD/DCoH: ptérine-4α-carbinolamine déshydratase; NBAT: neutral and basic amino acid transporter; Neuro-D1: neurogenic differentiation factor 1/β-cell E-box transactivator 2; Ins-1 et 2: gènes codant pour l’insuline; Glut2: glucose transporter 2 ; 1,3-BGD : glycéraldéhyde-3-phosphate déshydrogénase ; IAPP : islet amyloid poly-peptide; GCK: glucokinase; IPF-1: insulin promoter factor 1. |
D’identification plus récente, le MODY-5 dû à des mutations du gène codant pour HNF-1β est associé à un phénotype complexe, avec atteinte multiviscérale. On observe souvent chez les porteurs de ces mutations, outre le diabète, des anomalies rénales, morphologiques (kystes rénaux, anomalies du développement rénal) et fonctionnelles avec une insuffisance rénale, une atrophie pancréatique et des anomalies des voies génitales et du bilan hépatique [19]. Ce type de MODY semble plus fréquent que ne le laisse penser le petit nombre des observations rapportées dans la littérature.
Le facteur de transcription IPF-1 (insulin promoter factor-1) joue un rôle fondamental à la fois dans le développement embryonnaire du pancréas et, plus tard, dans le contrôle de la transcription de gènes pancréatiques spécifiques de tissus (Tableau I) (→). IPF-1 est normalement exprimé dans toutes les cellules du bourgeon pancréatique. Son absence chez des souris dont le gène codant pour IPF-1 a été invalidé conduit à l’arrêt du développement pancréatique au stage de bourgeon et à l’agénésie pancréatique. Le MODY-4 par mutation d’IPF-1 semble très rare; la mutation homozygote de ce gène était associée dans une famille à une agénésie pancréatique responsable d’un diabète néonatal et d’une insuffisance pancréatique exocrine [20]. Les sujets porteurs de la mutation à l’état hétérozygote avaient un diabète clinique- ment non insulinodépendant, secondaire à des altérations profondes de l’insulinosécrétion.
(→) m/s 2002, n°4, p. 467
Le facteur de transcription Neuro-D1 (aussi connu comme β-2) est impliqué dans le développement du pancréas endocrine. Son absence chez des souris dont le gène codant pour Neuro-D1 a été invalidé est associée à une diminution de la masse de cellules β-pancréatiques, à une diminution de l’expression du gène codant pour l’insuline et à un diabète sévère. Chez l’homme, le MODY- 6 par mutation de Neuro-D1 semble également très rare et ses manifestations cliniques peu décrites.
Conclusions
La découverte des gènes du MODY a considérablement fait avancer la compréhension des mécanismes moléculaires de l’homéostasie glycémique, notamment ceux impliqués dans la sécrétion d’insuline. De même, cela a permis de mieux définir les phénotypes cliniques de ces diabètes. On peut ainsi distinguer une forme de diabète proche par ses caractéristiques du diabète de type 2 de l’adulte (MODY-1 et MODY-3) et une forme identifiable très tôt et relativement bénigne (MODY-2). Le démembrement des MODY se poursuit et les études reliant les anomalies moléculaires aux altérations fonctionnelles sont indispensables pour mieux comprendre la physiopathologie de ces affections. À ce jour, les variants des gènes de MODY ne semblent pas jouer un rôle majeur dans la physiopathologie du diabète de type 2 dans sa forme commune. Cependant, l’identification de ces gènes a ouvert de nouvelles perspectives dans la compréhension des mécanismes de la différenciation et de la fonction des îlots pancréatiques. On peut également en attendre une amélioration de la prise en charge des patients fondée sur un pronostic plus précis et, à terme, sur une thérapeutique adaptée aux particularités de ces syndromes.
Références
- Fajans SS, Bell GI, Polonsky KS. Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N Engl J Med 2001; 345 : 971–80. [Google Scholar]
- Timsit J, Dubois-Laforgue D, Boitard C, Bellané- Chantelot C, Velho G. Physiopathologie, présentation clinique et complications des diabètes de type MODY. In: Actualités néphrologiques. Paris: Flammarion Médecine-Sciences, 2002: 89–98. [Google Scholar]
- Velho G, Froguel P, Clément K, et al. Primary pancreatic beta-cell secretory defect caused by mutations in the glucokinase in kindreds of maturity onset diabetes of the young. Lancet 1992; 340: 444–8. [Google Scholar]
- Byrne MM, Sturis J, Clément K, et al. Insulin secretory abnormalities in subjects with hyperglycemia due to glucokinase mutations. J Clin Invest 1994; 93: 1120–30. [Google Scholar]
- Velho G, Petersen KF, Perseghin G, et al. Impaired hepatic glycogen synthesis in glucokinase-deficient (MODY-2) subjects. J Clin Invest 1996; 98: 1755–61. [Google Scholar]
- Hattersley AT, Beards F, Ballantyne E, et al. Mutations in the glucokinase gene of the fetus result in reduced birth weight. Nat Genet 1998; 19: 268–70. [Google Scholar]
- Velho G, Blanché H, Vaxillaire M, et al. Identification of 14 new glucokinase mutations and description of the clinical profile of 42 MODY-2 families. Diabetologia 1997; 40: 217–24. [Google Scholar]
- Pearson ER, Velho G, Clark P, et al. β-cell genes and diabetes: quantitative and qualitative differences in the pathophysiology of hepatic nuclear factor 1- alpha and glucokinase mutations. Diabetes 2001; 50 (suppl 1): S101–7. [Google Scholar]
- Velho G, Vaxillaire M, Boccio V, Charpentier G, Froguel P. Diabetes complications in NIDDM kindreds linked to the MODY-3 locus on chromosome 12q. Diabetes Care 1996; 19: 915–9. [Google Scholar]
- Byrne MM, Sturis J, Menzel S, et al. Altered insulin secretory responses to glucose in diabetic and nondiabetic subjects with mutations in the diabetes mellitus susceptibility gene MODY on chromosome 12. Diabetes 1996; 45: 1503- 10. [Google Scholar]
- Pontoglio M, Sreenan S, Roe M, et al. Defective insulin secretion in hepatocyte nuclear factor 1-α deficient mice. J Clin Invest 1998; 101: 2215–22. [Google Scholar]
- Dukes ID, Sreenan S, Roe MW, et al. Defective pancreatic b-cell glycolytic signaling in Hepatocyte Nuclear Factor-1α- deficient mice. J Biol Chem 1998; 273: 24457–64. [Google Scholar]
- Vaxillaire M, Abderrahmani A, Boutin P, et al. Anatomy of a homeoprotein revealed by the analysis of human MODY3 mutations. J Biol Chem 1999; 274: 35639–46. [Google Scholar]
- Pontoglio M, Prié D, Cheret C, et al. HNF-1α controls renal glucose reabsorption in mouse and man. EMBO Rep 2000; 1: 359–65. [Google Scholar]
- Isomaa B, Henricsson M, Lehto M, et al. Chronic diabetic complications in patients with MODY3 diabetes. Diabetologia 1998; 41: 467–73. [Google Scholar]
- Stoffel M, Duncan SA. The maturity-onset diabetes of the young (MODY1) transcription factor HNF4 alpha regulates expression of genes required for glucose transport and metabolism. Proc Natl Acad Sci USA 1997; 94: 13209–14. [Google Scholar]
- Thomas H, Jaschkowitz K, Bulman M, et al. A distant upstream promoter of the HNF-4alpha gene connects the transcription factors involved in maturity-onset diabetes of the young. Hum Mol Genet 2001; 10: 2089–97. [Google Scholar]
- Shih DQ, Screenan S, Munoz KN, et al. Loss of HNF-1 alpha function in mice leads to abnormal expression of genes involved in pancreatic islet development and metabolism. Diabetes 2001; 50: 2472–80. [Google Scholar]
- Lindner TH, Njolstad PR, Horikawa Y, et al. A novel syndrome of diabetes mellitus, renal dysfunction and genital malformation associated with a partial deletion of the pseudo-POU domain of hepatocyte nuclear factor-1 beta. Hum Mol Genet 1999; 8: 2001–8. [Google Scholar]
- Stoffers DA, Ferrer J, Clarke WL, Habener JF. Early-onset type 2 diabetes mellitus (MODY4) linked to IPF1. Nat Genet 1997; 17: 138–9. [Google Scholar]
Liste des tableaux
Sous-types de MODY. * Distribution selon les populations ; Tm : taux maximal de réabsorption ; HNF : hepatocyte nuclear factor; IPF-1 : insulin promoter factor 1 ; Neuro-D1/β2 : neurogenic differentiation factor 1/β-cell E-box transactivator 2 ; Glut2 : glucose transporter 2 ; L-PK : liver-specific pyruvate kinase ; AldoB : aldolase B ; 1,3-BGD : glycéraldéhyde-3-phosphate déshydrogénase ; SGLT2 : sodium glucose transporter 2 ; NBAT : neutral and basic amino acid transporter ; PCD/DCoH : ptérine-4α-carbinolamine déshydratase ; IAPP : islet amyloid polypeptide.
Liste des figures
|
Figure 1. Représentation schématique du rôle régulateur des gènes du MODY dans l’homéostasie glycémique. A. Implication dans la sécrétion d’insuline en réponse au glucose: (a) phosphorylation du glucose en G-6-P par la glucokinase; (b) production d’ATP par la glycolyse et par le cycle de Krebs; (c) augmentation du rapport ATP/ADP intracellulaire ; (d) fermeture des canaux potassiques ATP-dépendants; (e) augmentation de la concentration intracellulaire de K+; (f) dépolarisation de la membrane cellulaire; (g) ouverture des canaux calciques dépendants du voltage ; (h) augmentation de la concentration intracellulaire de Ca2+ ; (i) exocytose de l’insuline. B. Implication de la glucokinase dans la synthèse postprandiale de glycogène hépatique. |
| Dans le texte | |
|
Figure 2. Représentation schématique du réseau interconnectant les facteurs de transcription impliqués dans le MODY et leurs gènes cibles. Les flèches représentent des régulations transcriptionelles positives ou négatives dans le cas de l’effet de SHP-1 sur HNF4α. HNF-1α et HNF-1β forment des homo- et des hétérodimères. HNF: hepatocyte nuclear factor ; SHP-1: small heterodimer partner 1; AldoB: aldolase B; LPK: liver-specific-pyruvate kinase; PCD/DCoH: ptérine-4α-carbinolamine déshydratase; NBAT: neutral and basic amino acid transporter; Neuro-D1: neurogenic differentiation factor 1/β-cell E-box transactivator 2; Ins-1 et 2: gènes codant pour l’insuline; Glut2: glucose transporter 2 ; 1,3-BGD : glycéraldéhyde-3-phosphate déshydrogénase ; IAPP : islet amyloid poly-peptide; GCK: glucokinase; IPF-1: insulin promoter factor 1. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.