")
")
| Issue |
Med Sci (Paris)
Volume 19, Number 8-9, Août-Septembre 2003
|
|
|---|---|---|
| Page(s) | 840 - 846 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/20031989840 | |
| Published online | 15 August 2003 | |
Hyperglycémie et angiogenèse
Hyperglycemia and angiogenesis
Inserm U.36, Chaire de Médecine Expérimentale, Collège de France, 11, place Marcelin Berthelot, 75231 Paris Cedex 05, France, et Service de Diabétologie, Hôpital Bichat, Paris, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
Les complications vasculaires, conséquences de l’hyperglycémie chronique, sont responsables de l’essentiel de la morbidité et de la mortalité chez les patients diabétiques. La cible principale de la toxicité chronique de l’hyperglycémie est la cellule endothéliale vasculaire. L’ischémie secondaire aux lésions vasculaires déclenche une réponse angiogénique. Chez le diabétique, cette réponse angiogénique à l’ischémie diffère selon le tissu en cause, excessive dans certains organes comme la rétine, elle est le plus souvent déficiente ailleurs. Les effets directs de l’hyperglycémie sur l’expression des facteurs de croissance vasculaires sont diversement appréciés, et pourraient varier en fonction de l’organe étudié. De plus, les effets délétères de l’hyperglycémie sur l’angiogenèse ne se limitent probablement pas aux effets directs sur l’expression de facteurs de croissance.
Abstract
Vascular complications of chronic hyperglycemia cause most of diabetes-associated morbidity and mortality. Main targets of chronic hyperglycemia are vascular endothelial cells. Ischemia is the late consequence of vascular damage in patients with diabetes and triggers an angiogenic response. In patients with diabetes, the angiogenic response to chronic ischemia can be excessive in some of the target organs and insufficient in others, in the same individual. The direct effects of hyperglycemia on the expression level of vascular growth factors have been variably appreciated and depend on the studied organ and model. Beyond the described effects of hyperglycemia on the expression level of vascular growth factors, direct and indirect effects of hyperglycemia on endothelial cell proliferation, extracellular matrix and metalloproteases might be involved in the pathology of angiogenesis in diabetes.
© 2003 médecine/sciences - Inserm / SRMS
L’hyperglycémie chronique, caractéristique des syndromes diabétiques, est à l’origine d’altérations dans la structure et la fonction des microvaisseaux et des macrovaisseaux. Ces lésions vasculaires sont responsables de l’essentiel de la morbidité et de la mortalité observées chez les diabétiques. Les altérations des microvaisseaux sont en cause dans la rétinopathie diabétique, première cause de cécité chez l’adulte jeune en Occident, de la néphropathie diabétique, première cause de mise en hémodialyse pour insuffisance rénale terminale, et de la neuropathie, première cause d’amputation non traumatique. Les lésions des gros vaisseaux, responsables d’infarctus du myocarde, d’accidents vasculaires cérébraux et d’ischémie des membres inférieurs, se distinguent chez les diabétiques par leur précocité et leur sévérité. Les altérations des vaisseaux sanguins chez les diabétiques sont diffuses : à titre d’exemple, la Figure 1 - coupe histologique d’un muscle de patient diabétique - montre des altérations des capillaires, qui sont occlus, et des artérioles, dont la paroi est épaissie, ce qui réduit la lumière du vaisseau (Figure 1). Les altérations vasculaires entraînent une ischémie tissulaire. Chez l’adulte, l’ischémie déclenche une série de processus visant à augmenter le flux sanguin dans le territoire lésé et qui permettent, par l’ouverture de réseaux collatéraux de suppléance et le développement de nouveaux vaisseaux, de prévenir la mort tissulaire. Ces processus hautement contrôlés d’angiogenèse et d’artériogenèse, dont les moteurs principaux sont l’hypoxie et l’hypoglycémie, paraissent pathologiques chez les diabétiques: excessifs au niveau de la rétine, ils sont responsables de la forme la plus sévère de rétinopathie; défectueux au niveau des gros vaisseaux, ils contribuent à la sévérité particulière des cardiopathies ischémiques chez les diabétiques. Cette dualité de réponse à l’ischémie chez les diabétiques pose un problème au médecin: si une réponse angiogénique forte est désirable au niveau des artères coronaires, une telle réponse est redoutée au niveau de la rétine [1]. De même, si des thérapeutiques à visée anti-angiogénique ont montré leur efficacité dans des modèles animaux de rétinopathie proliférante, de tels traitements, administrés pour traiter ou prévenir les rétinopathies diabétiques proliférantes, pourraient aggraver le pronostic vasculaire chez les diabétiques. Il est maintenant bien établi que le risque de complication vasculaire est, chez le diabétique, proportionnel au niveau glycémique moyen et à la durée d’exposition à l’hyperglycémie [2–4]. Si les mécanismes cellulaires de la toxicité de l’hyperglycémie chronique commencent à être compris, ses cibles restent encore incertaines. Après une brève présentation des mécanismes cellulaires activés par l’hyperglycémie chronique, cet article présentera ce que l’on sait des cibles de l’hyperglycémie chronique et des mécanismes qui pourraient expliquer la pathologie de l’angiogenèse chez les diabétiques.
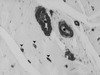 |
Figure 1. Pathologie vasculaire chez les sujets diabétiques. Biopsie musculaire en tissu « sain » chez un diabétique ayant une ischémie critique des membres inférieurs (marquage avec un anticorps anti-actine). Athérosclérose marquée sur les artérioles: épaississement de la paroi vasculaire responsable d’une réduction de la lumière vasculaire (*) ; occlusions capillaires (pointes de flèche rouges). |
Toxicité de l’hyperglycémie chronique
Mécanismes cellulaires impliqués
Quatre voies intracellulaires paraissent activées par l’hyperglycémie: le flux de la voie des polyols et de la voie des hexosamines est augmenté, la production de produits de glycation avancée (AGE) est augmentée et certaines isoformes de la protéine kinase C (PKC) sont activées via une augmentation de la production de diacyl- glycérol (DAG). Dans certains types cellulaires, l’hyperglycémie active également le facteur de transcription NF-κB. Il a récemment été montré que, dans des cellules endothéliales en culture, l’activation de toutes ces voies est la conséquence commune d’une augmentation de la production de l’ion superoxyde 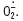 Ce stress oxydant est lui-même secondaire à un emballement du cycle de Krebs qui dépasse alors les capacités de la chaîne respiratoire mitochondriale [5]. Ainsi, les voies alternes de métabolisme du glucose sont activées par emballement de la glycolyse, mais aussi par l’inhibition partielle, par l’ion superoxyde de la glycéraldéhyde- 3-phosphate déshydrogénase (GAPDH) [6].
Ce stress oxydant est lui-même secondaire à un emballement du cycle de Krebs qui dépasse alors les capacités de la chaîne respiratoire mitochondriale [5]. Ainsi, les voies alternes de métabolisme du glucose sont activées par emballement de la glycolyse, mais aussi par l’inhibition partielle, par l’ion superoxyde de la glycéraldéhyde- 3-phosphate déshydrogénase (GAPDH) [6].
Perturbations des flux sanguins régionaux chez les diabétiques
L’analyse des données de la littérature concernant les flux sanguins régionaux chez les diabétiques est délicate, car les résultats des études sont souvent contradictoires. Hors contexte diabétique, le flux sanguin rétinien total est augmenté par une hyperglycémie aiguë. Chez les diabétiques sans rétinopathie, le flux sanguin rétinien est diminué en hyperglycémie chronique, mais une élévation de la glycémie est capable, comme chez les témoins, d’augmenter le flux sanguin rétinien total. En revanche, le flux sanguin rétinien total augmente dans les formes sévères de rétinopathie, en proportion de l’ischémie rétinienne. La réduction du flux sanguin dans un contexte d’hyperglycémie chronique dépend probablement en partie de l’activation de la PKC, puisqu’un inhibiteur sélectif de la PKC est capable de d’augmenter le flux sanguin rétinien [7].
La participation de facteurs hémodynamiques est bien démontrée pour la néphropathie diabétique et, d’une manière plus générale, la démonstration que l’inhibition du système rénine-angiotensine est capable de ralentir la progression des lésions vasculaires chez les diabétiques est un fort argument en faveur de la participation de facteurs hémodynamiques à la physiopathologie des complications de micro- et de macroangiopathies. Toutefois, il convient de noter que les inhibiteurs de l’enzyme de conversion, qui ont démontré leur supériorité par rapport aux autres agents antihypertenseurs pour la prévention des complications vasculaires chez les diabétiques, ont également une action anti-oxydante significative [8].
Cibles de l’hyperglycémie chronique
La nature des cibles de l’hyperglycémie chronique est encore incertaine. Si les cellules endothéliales sont les cibles les mieux caractérisées, d’autres types cellulaires participent à la physiopathologie des complications vasculaires chez les diabétiques.
Bien qu’elles soient les premières exposées à l’hyperglycémie intravasculaire, les cellules endothéliales vasculaires sont incapables, à la différence des cellules musculaires lisses vasculaires, de réduire leur capture de glucose lorsqu’elles sont exposées à des concentrations excessives de glucose, malgré l’existence d’une régulation négative, comme dans une majorité des tissus, de l’expression du transporteur de glucose ubiquitaire GLUT1. La plupart des anomalies biochimiques décrites dans des cellules exposées à une hyperglycémie chronique l’ont été dans des cellules endothéliales. Des anomalies précoces de la barrière endothéliale sont bien caractérisées, et se manifestent par une fuite de plasma dans l’espace périvasculaire, en partie modulée par la carence en insuline [9]. Le diabète est par ailleurs caractérisé par un dysfonctionnement endothélial généralisé, avec une altération des fonctions anticoagulante et anti-inflammatoire de l’endothélium et une diminution de la vasodilatation dépendant de l’endothélium.
Néanmoins, la première anomalie histologique observée chez les diabétiques est la perte des péricytes, qui constituent la couche unique de cellules contractiles au niveau des capillaires. En plus de leurs propriétés contractiles, les péricytes participent à l’élaboration de la matrice extracellulaire. Bien qu’il ait été montré que, in vitro, les cellules musculaires lisses, auxquelles les péricytes sont apparentés, sont capables de régler leur transport de glucose face à une hyperglycémie, il apparaît que les péricytes sont plus sensibles que les cellules endothéliales aux dégâts de l’hyperglycémie. Ils apparaissent ainsi plus sensibles au stress oxydant [10]. Les péricytes des diabétiques répondent moins bien au NO et à l’endothéline-1, dont la synthèse est augmentée. Les cellules sanguines circulantes sont probablement aussi en cause dans la physiopathologie des lésions vasculaires chez les diabétiques. L’adhérence des leucocytes est ainsi favorisée par l’expression de molécules d’adhésion par les cellules endothéliales, et il a été montré que les leucocytes ainsi piégés participent aux lésions des cellules endothéliales [11, 12].
Le rôle de lésions précoces des tissus non vasculaires émerge progressivement. Il semble ainsi que, au niveau de la rétine, les lésions des cellules gliales sont très précoces et pourraient participer, voire avoir un rôle causal, à la physiopathologie de la microangiopathie [10, 13].
Altération des processus d’angiogenése chez les diabétiques
Au-delà de l’aspect excessif de l’angiogenèse qui caractérise les formes les plus sévères de la rétinopathie et de la néphropathie diabétique, et qui contraste avec l’insuffisance, chez les diabétiques, des processus d’artériogenèse au niveau des vaisseaux coronaires ou des membres inférieurs, plusieurs autres modèles de diabète décrivent une angiogenèse défectueuse, le plus souvent déficiente : angiogenèse embryonnaire, ou accompagnant les processus inflammatoires et la cicatrisation, ou angiogenèse de revascularisation, après greffe d’organes avasculaires tels que les îlots de Langerhans [14].
L’angiogenèse chez l’adulte, activée dans des circonstances physiologiques telles que la maturation du corps jaune ovarien et de l’endomètre au cours du cycle menstruel, par exemple, est aussi un élément important des processus inflammatoires et cicatriciels, et un déterminant essentiel de l’évolution des tumeurs solides [15]. L’angiogenèse constitue l’un des mécanismes de formation de nouveaux vaisseaux chez l’adulte, par bourgeonnement à partir de vaisseaux déjà existants. Elle aboutit souvent, en dehors des circonstances où elle est physiologique, et probablement parce qu’elle est alors imparfaitement régulée, à la formation de vaisseaux de petit calibre et peu fonctionnels, tels les néovaisseaux rétiniens observés chez les diabétiques, mais aussi dans d’autres formes de rétinopathie avec ischémie rétinienne [15, 16]. Le remodelage de vaisseaux préexistants par artériogenèse peut conduire au développement de vaisseaux collatéraux fonctionnels en quelques semaines [17]. Ce remodelage proximal, en amont de la zone ischémique, s’associe à une angiogenèse distale dans la plupart des cas.
Déclenchement et mécanismes de l’angiogenése
Les événements déclenchant les processus angiogéniques sont maintenant bien caractérisés. Un rôle central est reconnu au stress métabolique (baisse de la PO2, baisse du pH, hypoglycémie), qui s’exprime en grande partie par la régulation des facteurs de transcription sensibles à l’hypoxie HIF (hypoxia-inducible factor) [18], et aux facteurs mécaniques tels qu’une variation de pression ou de flux. Enfin, dans la plupart des circonstances, une inflammation locale paraît nécessaire [15, 16].
L’angiogenèse nécessite l’intervention coordonnée de multiples facteurs qui interviennent dans les différentes étapes aboutissant à la formation d’un nouveau vaisseau. La séquence d’événements mis en jeu peut être ainsi résumée: vasodilatation et déstabilisation de la paroi vasculaire; dégradation de la matrice extracellulaire; prolifération et migration des cellules endothéliales, ou incorporation dans le vaisseau en bourgeonnement de précurseurs d’angioblastes circulants issus de la moelle osseuse; établissement des contacts intercellulaires pour former un tube; recrutement et prolifération de péricytes/cellules musculaires lisses vasculaires. Les principales molécules impliquées à chacune de ces étapes sont présentées sur la Figure 2.
 |
Figure 2. Étapes de l’angiogenèse et principaux facteurs de croissance impliqués. La figure schématise les principales étapes de l’angiogenèse; sous chaque étape sont indiqués les principaux facteurs impliqués. EGF: epithelial growth factor; bFGF: basic fibroblast growth factor; HGF: hepatocyte growth factor; IGF1: insulin-like growth factor 1; MMP: métalloprotéases de la matrice extracellulaire; PDGF: platelet derived growth factor; PLGF: placental growth factor; TGF-β: transforming growth factor β ; TNF- α: tumor necrosis factorα; VEGF: vascular endothelial growth factor (d’après [35]). |
Perturbations de l’angiogenése chez les diabétiques : aspects moléculaires et cellulaires
De multiples altérations cellulaires et moléculaires ont été décrites, dans des conditions d’hyperglycémie in vitro, dans des modèles de diabète chez des rongeurs ou encore chez des patients diabétiques. Ces altérations sont toutes susceptibles de participer aux anomalies de l’angiogenèse observées chez les diabétiques, mais pas toujours dans le même sens.
Défaut de mobilisation des angioblastes
Il a récemment été montré que les angioblastes, cellules précurseurs des cellules endothéliales issues de la moelle osseuse, que l’on peut caractériser par l’expression des marqueurs CD34 et VEGF-R2, sont mobilisables par des cytokines, dont le VEGF (vascular endothelial growth factor), et s’incorporent dans les vaisseaux en cours de développement chez l’adulte [19, 20]. Or des défauts de mobilisation et de fonction des progéniteurs endothéliaux ont récemment été décrits au cours du diabète [21, 22].
Anomalies de survie, de prolifération et de fonction des cellules endothéliales
Les cellules endothéliales exposées à l’hyperglycémie ont une apoptose augmentée, ainsi que des défauts de prolifération et d’entrée dans le cycle cellulaire. Ces anomalies sont probablement en grande partie la conséquence d’un stress oxydant, et on conçoit que ces dysfonctionnement puissent contribuer aux défauts de l’angiogenèse chez les diabétiques
Anomalies de sécrétion du VEGF
Le VEGF est le principal facteur de croissance vasculaire. Essentiel pendant le développement embryonnaire, puisque l’expression d’un seul des deux allèles n’est pas suffisante pour permettre le développement du réseau vasculaire, il possède aussi un rôle majeur dans l’angiogenèse physiologique et pathologique chez l’adulte. Le VEGF agit à tous les stades de l’angiogenèse: il augmente la perméabilité vasculaire, favorise la prolifération et la migration des cellules endohéliales et mobilise les angioblastes. C’est enfin un agent chimiotactique pour les monocytes et les cellules musculaires lisses vasculaires. L’expression du VEGF est réglée par les facteurs sensibles à l’hypoxie et par plusieurs cytokines et facteurs de croissance (TGF [transforming growth factor]-β, IL [interkeukine]-1, FGF [fibroblast growth factor], PDGF [platelet-derived growth factor], IGF [insulin-like growth factor]-1…).
Un rôle important dans la physiopathologie des complications microvasculaires, en particulier rétiniennes, du diabète est attribué à un dérèglement de l’expression du VEGF [23]. Dans la rétine, le VEGF peut être exprimé par plusieurs types de cellules: cellules de l’épithélium pigmentaire rétinien, cellules gliales, cellules ganglionnaires, péricytes et cellules endothéliales. Il a été observé une surexpression précoce du VEGF, après 8 jours d’hyperglycémie chez la souris, sous la dépendance de HIF-1α [9]. Chez le primate, des injections répétées de VEGF sont capables de reproduire les anomalies vasculaires observées chez les diabétiques.
La surexpression de VEGF observée en cas d’hyperglycémie pourrait être la conséquence directe de l’activation de la PKC, ou une conséquence de la réduction des flux sanguins, responsables d’une ischémie rétinienne [24]. Il est possible que la surexpression du VEGF soit un mécanisme de défense des cellules neurales de la rétine contre l’apoptose induite par l’hyperglycémie [13], ou la conséquence d’un défaut d’expression de l’angiopoïétine 1 [25] ou de la surexpression du TGF-β. Elle pourrait, enfin, être sous la dépendance de l’angiotensine II [26].
Néanmoins, les données suggérant un rôle pathologique d’une surexpression précoce du VEGF dans la rétine contrastent avec plusieurs faits. Les modèles où une surexpression précoce du VEGF a été démontrée sont des modèles où l’on n’observe pas de rétinopathie proliférante en réponse à l’hyperglycémie. Par ailleurs, si l’on peut attribuer au VEGF
un rôle important dans les phases précoces de la maladie, faites d’augmentation de perméabilité capillaire, ou dans les phases tardives, faites de prolifération vasculaire, plus de 10 ans en général séparent ces deux phases chez l’homme, et la rétinopathie proliférante ne s’observe qu’en réponse à une ischémie rétinienne considérable. Enfin, plusieurs modèles décrivent plutôt un défaut d’expression du VEGF, en particulier en réponse à une ischémie du membre inférieur [27]. Dans ce contexte, un travail récent a mis en évidence un défaut de production de VEGF par les fibroblastes de souris diabétiques [28]. Il a cependant été montré qu’il existe une régulation différentielle de l’expression du VEGF, de sorte qu’elle peut être augmentée par l’hyperglycémie au niveau de la rétine et diminuée au niveau du myocarde [29].
Dans ces conditions, si des anomalies d’expression du VEGF accompagnent très certainement les phases tardives de la rétinopathie diabétique, il est difficile, à ce jour, d’attribuer un rôle prépondérant à la seule dérégulation, par l’hyperglycémie et la carence en insuline, de l’expression du gène du VEGF dans la physiopathologie des complications vasculaires chez le diabétique.
Anomalies de sécrétion des autres facteurs de croissance vasculaire
Des anomalies dans l’expression de plusieurs autres facteurs de croissance vasculaire ont également été observées en réponse à l’hyperglycémie ou au diabète, qui pourraient participer à la pathologie de l’angiogenèse chez les diabétiques. Ces anomalies concernent le FGF [30], l’HGF (hepatocyte growth factor) [31], l’angiopoïétine 1 [25], le PLGF (placental growth factor), le PEDF (pigment epithelium-derived factor), un inhibiteur rétinien de l’angiogenèse [32]. Le manque de coordination entre les différents facteurs de croissance vasculaire, certains surexprimés tandis que d’autres sont sous-exprimés, ou le manque de coordination dans le temps joue probablement un rôle dans l’angiogenèse pathologique chez les diabétiques. À titre d’exemple, il a été montré dans des modèles de souris transgéniques que la surexpression du VEGF aboutit à la formation d’un réseau vasculaire exubérant, mais de mauvaise qualité, caractérisé par des défauts majeurs de perméabilité, anomalies que corrige la coexpression de l’angiopoïétine 1 [33]. De même, l’angiopoïétine 1 est capable, dans un modèle d’hyperglycémie expérimentale chez le rongeur, de corriger les altérations vasculaires précoces (défaut de perméabilité vasculaire, apoptose, adhésion des leucocytes) induits par la surexpression du VEGF [25].
Anomalies de la matrice extracellulaire
La matrice extracellulaire des vaisseaux est altérée chez les patients diabétiques. L’augmentation de l’épaisseur des membranes basales est une anomalie précoce et constante chez les diabétiques, et l’hyperglycémie augmente la synthèse de multiples composants de la matrice extracellulaire. Ces effets sont la conséquence de l’activation de la PKC, de l’expression du TGF-β ou de phénomènes mécaniques liés à l’augmentation des forces de cisaillement. Dans les phases tardives de la maladie, les altérations de la matrice extracellulaire sont majorées par la glycation non enzymatique. L’expression des gènes codant pour la fibronectine et pour divers collagènes, dont le collagène de type IV, principal composant des membranes basales, sont affectées par l’hyperglycémie [10]. En plus des anomalies quantitatives et qualitatives de la matrice extracellulaire, il semble que l’expression des protéases de la matrice extracellulaire, nécessaires aux étapes précoces de l’angiogenèse, soit réduite chez les diabétiques [34].
Rôle des produits de glycation avancée (AGE)
Les produits de glycation avancée, dont la production est augmentée chez les diabétiques, ont la capacité de modifier la structure et la fonction des protéines à longue demi-vie. C’est le cas en particulier des protéines de la matrice extracellulaire, qui deviennent ainsi moins digestibles par les protéases. Outre cet effet direct, les produits de glycation avancée sont impliqués à des niveaux multiples dans la physiopathologie des complications vasculaires chez les diabétiques, en raison notamment de leur capacité à majorer le stress oxydant des cellules endothéliales, ou de leur capacité à provoquer la sécrétion de cytokines par les cellules monocytaires, après liaison à des récepteurs.
Conclusions
Nous avons souhaité montrer la complexité de l’analyse des effets du diabète sur l’angiogenèse, et les implications de ces effets sur l’évolution des complications vasculaires. La poursuite des études des interactions hyperglycémie/angiogenèse devra prendre en compte l’hétérogénéité des cellules endothéliales, les interactions entre cellules endothéliales et cellules circulantes, entre cellules endothéliales et matrice extracellulaire, entre les facteurs de croissance vasculaires et leurs inhibiteurs, et l’orchestration fine des moments de leur entrée en action au cours des événements conduisant à la formation des nouveaux vaisseaux et à l’ouverture des vaisseaux collatéraux. Les futurs essais thérapeutiques de stimulation/inhibition de l’angiogenèse ne se limiteront probablement pas à l’administration, respectivement, d’un seul facteur de croissance ou d’un inhibiteur, mais feront probablement appel à des stratégies d’administration coordonnée de plusieurs des acteurs de l’angiogenèse.
Références
- Duh E, Aiello LP. Vascular endothelial growth factor and diabetes. The agonist versus antagonist paradox. Diabetes 1999; 48: 1899–906. [Google Scholar]
- The diabetes control and complication trial group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulindependent diabetes mellitus. N Engl J Med 1999; 329: 977–87. [Google Scholar]
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood glucose control with sulfonyureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998; 352: 837–53. [Google Scholar]
- Stratton IM, Adler AI, Neil HA, et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. Br Med J 2000; 321: 405–12. [Google Scholar]
- Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001; 414: 813–20. [Google Scholar]
- Hammes HP, Du X, Edelstein D, et al. Benfotiamine blocks three major pathways of hyperglycemic damage and prevents experimental diabetic retinopathy. Nat Med 2003; 9: 294–9. [Google Scholar]
- Ishii H, Jirousek MR, Koya D, et al. Amelioration of vascular dysfunction in diabetic rats by an oral PKC β inhibitor. Science 1996; 272: 728–31. [Google Scholar]
- Munzel T, Keaney JF, Jr. Are ACE inhibitors a «magic bullet» against oxidative stress ? Circulation 2001; 104: 1571–4. [Google Scholar]
- Poulaki V, Qin W, Joussen AM, et al. Acute intensive insulin therapy exacerbates diabetic blood-retinal barrier breakdown via hypoxia-inducible factor- 1alpha and VEGF. J Clin Invest 2002; 109: 805–15. [Google Scholar]
- Lorenzi M, Gerhardinger C. Early cellular and molecular changes induced by diabetes in the retina. Diabetologia 2001; 44: 791–804. [Google Scholar]
- Joussen AM, Murata T, Tsujikawa A, Kirchhof B, Bursell SE, Adamis AP. Leukocyte-mediated endothelial cell injury and death in the diabetic retina. Am J Pathol 2001; 158: 147–52. [Google Scholar]
- Joussen AM, Poulaki V, Mitsiades N, et al. Suppression of Fas-FasLinduced endothelial cell apoptosis prevents diabetic blood-retinal barrier breakdown in a model of streptozotocin-induced diabetes. FASEB J 2003; 17: 76–8. [Google Scholar]
- Barber AJ. A new view of diabetic retinopathy: a neurodegenerative disease of the eye. Prog Neuropsychopharmacol Biol Psychiatry 2003; 27: 283–90. [Google Scholar]
- Martin A, Komada MR, Sane DC. Abnormal angiogenesis in diabetes mellitus. Med Res Rev 2003; 23: 117–45. [Google Scholar]
- Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature 2000; 407: 249–57. [Google Scholar]
- Yancopoulos GD, Davis S, Gale NW, Rudge JS, Wiegand SJ, Holash J. Vascularspecific growth factors and blood vessel formation. Nature 2000; 407: 242–8. [Google Scholar]
- Schaper W, Ito WD. Molecular mechanisms of coronary collateral vessel growth. Circ Res 1996; 79: 911–9. [Google Scholar]
- Semenza G. HIF-1, O(2), and the 3 PHDs: how animal cells signal hypoxia to the nucleus. Cell 2001; 107: 1–3. [Google Scholar]
- Asahara T, Murohara T, Sullivan A, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science 1997; 275: 964–7. [Google Scholar]
- Asahara T, Masuda H, Takahashi T, et al. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ Res 1999; 85: 221–8. [Google Scholar]
- Schatteman GC, Hanlon HD, Jiao C, Dodds SG, Christy BA. Blood-derived angioblasts accelerate blood-flow restoration in diabetic mice. J Clin Invest 2000; 106: 571–8. [Google Scholar]
- Tepper OM, Galiano RD, Capla JM, et al. Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation 2002; 106: 2781–6. [Google Scholar]
- Aiello LP, Wong JS. Role of vascular endothelial growth factor in diabetic vascular complications. Kidney Int 2000; 77 (suppl): S113–9. [Google Scholar]
- Koya D, King GL. Protein kinase C activation and the development of diabetic complications. Diabetes 1998; 47: 859–66. [Google Scholar]
- Joussen AM, Poulaki V, Tsujikawa A, et al. Suppression of diabetic retinopathy with angiopoietin-1. Am J Pathol 2002; 160: 1683–93. [Google Scholar]
- Gilbert RE, Kelly DJ, Cox AJ, et al. Angiotensin converting enzyme inhibition reduces retinal overexpression of vascular endothelial growth factor and hyperpermeability in experimental diabetes. Diabetologia 2000; 43: 1360–7. [Google Scholar]
- Rivard A, Silver M, Chen D, et al. Rescue of diabetesrelated impairment of angiogenesis by intramuscular gene therapy with adeno-VEGF. Am J Pathol 1999; 154: 355–63. [Google Scholar]
- Lerman OZ, Galiano RD, Armour M, Levine JP, Gurtner GC. Cellular dysfunction in the diabetic fibroblast: impairment in migration, vascular endothelial growth factor production, and response to hypoxia. Am J Pathol 2003; 162: 303–12. [Google Scholar]
- Chou E, Suzuma I, Way KJ, et al. Decreased cardiac expression of vascular endothelial growth factor and its receptors in insulinresistant and diabetic states: a possible explanation for impaired collateral formation in cardiac tissue. Circulation 2002; 105: 373–9. [Google Scholar]
- Lowe WL Jr, Florkiewicz RZ, Yorek MA, Spanheimer RG, Albrecht BN. Regulation of growth factor mRNA levels in the eyes of diabetic rats. Metabolism 1995; 44: 1038. [Google Scholar]
- Morishita R, Aoki M, Nakamura S, et al. Potential role of a novel vascular modulator, hepatocyte growth factor (HGF), in cardiovascular disease: characterization and regulation of local HGF system. J Atheroscler Thromb 1997; 4: 12–9. [Google Scholar]
- Spranger J, Osterhoff M, Reimann M, et al. Loss of the antiangiogenic pigment epithelium-derived factor in patients with angiogenic eye disease. Diabetes 2001; 50: 2641–5. [Google Scholar]
- Thurston G, Suri C, Smith K, et al. Leakage-resistant blood vessels in mice transgenically overexpressing angiopoietin-1. Science 1999; 286: 2511–4. [Google Scholar]
- Portik-Dobos V, Anstadt MP, Hutchinson J, Bannan M, Ergul A. Evidence for a matrix metalloproteinase induction/activation system in arterial vasculature and decreased synthesis and activity in diabetes. Diabetes 2002; 51: 3063–8. [Google Scholar]
- Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med 2000; 6: 389–95. [Google Scholar]
Liste des figures
|
Figure 1. Pathologie vasculaire chez les sujets diabétiques. Biopsie musculaire en tissu « sain » chez un diabétique ayant une ischémie critique des membres inférieurs (marquage avec un anticorps anti-actine). Athérosclérose marquée sur les artérioles: épaississement de la paroi vasculaire responsable d’une réduction de la lumière vasculaire (*) ; occlusions capillaires (pointes de flèche rouges). |
| Dans le texte | |
|
Figure 2. Étapes de l’angiogenèse et principaux facteurs de croissance impliqués. La figure schématise les principales étapes de l’angiogenèse; sous chaque étape sont indiqués les principaux facteurs impliqués. EGF: epithelial growth factor; bFGF: basic fibroblast growth factor; HGF: hepatocyte growth factor; IGF1: insulin-like growth factor 1; MMP: métalloprotéases de la matrice extracellulaire; PDGF: platelet derived growth factor; PLGF: placental growth factor; TGF-β: transforming growth factor β ; TNF- α: tumor necrosis factorα; VEGF: vascular endothelial growth factor (d’après [35]). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.