")
")
| Issue |
Med Sci (Paris)
Volume 19, Number 6-7, Juin-Juillet 2003
|
|
|---|---|---|
| Page(s) | 668 - 670 | |
| Section | Le Magazine : Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/20031967668 | |
| Published online | 15 juin 2003 | |
À propos des neurodégénérescences associées à un déficit en pantothénate-kinase ou PKAN
About pantothenate kinaseassociated degeneration or PKAN
9, rue Basse, 54330 Clerey-sur-Brenon, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Les troubles neurologiques extra-pyramidaux avec augmentation du fer sérique se rencontrent au cours de trois maladies dont le défaut moléculaire a été identifié:
la neuroferritinopathie due à des mutations du gène codant pour la chaîne légère de la ferritine;
l’acéruléoplasminémie due à une mutation du gène codant pour la céruloplasmine;
le syndrome d’Hallervorden-Spatz dû à un déficit en pantothénate kinase. Comme nous le suggérions il y a quelques années (→), le temps semble venu de supprimer ces éponymes de sinistre mémoire pour appeler ce dernier syndrome: neurodégénérescence associée à un déficit en pantothénate kinase ou PKAN (→→).
(→) m/s 1997, n°1, p. 133
(→→) m/s 2002, n° 3, p. 280
Le syndrome d’Hallervorden-Spatz, une bien sombre histoire
Avant la Deuxième Guerre mondiale, au sein de l’hôpital de Brandebourg existait un asile d’aliénés. Il fut choisi pour devenir un des six centres d’élimination dans le cadre du programme d’euthanasie intitulé Aktion T- 4 du troisième Reich. Dès le début de la guerre, venant de toute l’Allemagne, de nombreux malades y furent transférés. Après une brève période d’observation, les « aliénés incurables », sous couvert d’être envoyés à la douche, étaient exterminés au monoxyde de carbone. En l’espace de deux ans (1939-1941), l’efficace programme Aktion T-4 fit périr ainsi 70273 personnes [1].
Une telle concentration de malades mentaux, avec « l’opportunité » de réaliser l’étude anatomo-pathologique des cerveaux dans d’excellentes conditions, n’a pas laissé Julius Hallervorden indifférent. Et - conscience professionnelle oblige - pour que le travail soit bien fait, il tint à examiner personnellement les malades et à prélever lui-même les cerveaux. C’est ainsi que furent publiés, pendant et après la guerre, des études concernant non seulement les dégénérescences cérébrales familiales, en particulier celle qui porte son nom, mais aussi concernant l’effet du monoxyde de carbone sur les cerveaux fœtaux. Ces publications accrurent la renommée du Kaiser Wilhem Institut où Julius Hallervorden conserva, jusqu’à sa retraite, un poste à responsabilité.
Le gène PANK2
La mise en cause du gène PANK2 put être faite grâce à l’étude d’une grande famille amish, qui permit de trouver un locus en 20p13 [2] et d’isoler un gène homologue du gène murin Pank1. Appelé PANK2, ce gène, qui appartient à une famille de 4 gènes PANK chez l’homme, code pour une pantothénate kinase, enzyme de régulation de la biosynthèse du Coenzyme A (CoA). Celui-ci catalyse la phosphorylation cytosolique du pantothénate (vitamine B5) et intervient aussi dans le métabolisme des acides gras. Chez la mouche et chez la levure, où il existe un seul gène Pank, les mutations entraînant l’absence de pantothénate kinase sont létales.
Une symptomatologie plus claire
Une étude récente, faite par l’équipe ayant isolé le gène PANK2, a rassemblé 123 malades provenant de 98 familles. Elle a permis de mieux définir la maladie et les relations génotype-phénotype [3].
Les formes typiques
Les premiers signes apparaissent généralement dans la première décennie de la vie. Ils se caractérisent par une dystonie atteignant d’abord les membres, puis apparaissent des troubles de la marche avec spasticité et déformation des pieds (varus équin). La rigidité s’accompagne de mouvements involontaires, de type choréoatéthosique. La survenue d’une rétinite pigmentaire n’est pas rare. Les nerfs crâniens sont touchés, ce qui entraîne une dysarthrie avec troubles de la déglutition. L’examen neurologique montre une hyperréflexie et un signe de Babinski.
L’IRM cérébrale révèle des images caractéristiques: le globus pallidus présente une hypodensité avec, dans la région médiane, une zone effilée d’hyperdensité donnant une image en « œil de tigre » spécifique de la maladie et permettant le diagnostic (Figure 1).
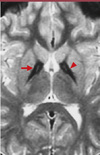 |
Figure 1. IRM chez un patient atteint de maladie d’Hallervorden-Spatz: image caractéristique en « œil de tigre » (reproduit de [5]) |
Une détérioration mentale s’installe progressivement et la mort survient en général avant 30 ans.
Les formes atypiques
À côté de ce tableau classique, il existe des formes à début plus tardif (2e ou 3e décennie) où la dysarthrie est souvent la première manifestation clinique. II existe aussi des troubles psychiatriques (palilalie1, dépression, psychose). Comme dans la maladie de Parkinson, on peut observer des blocages dans la marche lors de tournants ou d’inégalités du sol. La rétinopathie est plus rare, l’évolution plus lente, la perte de la marche ne survenant que 30 à 40 ans après la survenue des premiers signes (Tableau I).
Phénotype des neurodégénerescences associées à un déficit en pantothénate kinase.
Le syndrome HARP (hypopre β-lipoproteinemia, acantocytosis, retinitis pigmentosa and pallidal degeneration) comporte, outre les signes décrits cidessus, une anomalie des lipoprotéines plasmatiques (absence de fraction pré- β sur l’électrophorèse à haute résolution) et une acanthocytose [4]. L’image en œil de tigre est retrouvée, ainsi que des mutations du gène PANK2.
Relation génotype-phénotype
Des mutations du gène sont présentes dans tous les cas classés dans les formes typiques. Dans les cas atypiques, on les retrouve chez 35 % des malades. Ce sont souvent des mutations faux-sens, qui permettent de conserver une activité enzymatique résiduelle. Pour l’ensemble, deux mutations sont nettement plus fréquentes: Thr418Met et Gly411Arg. Mais cette dernière pose problème: dans 6 familles, en effet, elle est présente à l’état hétérozygote, aucune mutation n’ayant été retrouvée sur le deuxième allèle. L’hypothèse d’un effet semi-dominant pourrait être envisagée, mais semble peu probable, car l’étude des parents montre clairement que ceux-ci sont porteurs de la mutation G411R et qu’ils sont complètement asymptomatiques.
Conclusion
Dans cette neurodégénérescence due à une déficience en panthoténate kinase, le diagnostic peut être fait in vivo grâce à l’IRM cérébrale. Les relations génotype- phénotype deviennent de plus en plus claires, avec l’individualisation d’une forme atypique, à évolution plus lente et présentant des signes également présents dans la maladie de Parkinson. Mais dans cette forme, 65 % des cas ne sont pas dus à une atteinte du gène PANK2, et la quantité de fer cérébral peut être normale. Il reste donc d’autres causes à trouver. Enfin, des essais thérapeutiques par apports supplémentaires de vitamine B5 doivent être tentés dans les formes à début tardif pour voir s’ils sont capables de retarder l’apparition ou l’aggravation des symptômes.
Répétition involontaire du même mot ou de la même phrase.
Références
- Shevell M. Hallervorden and History. N Engl J Med 2003; 348: 3–4. [Google Scholar]
- Zhou B, Westaway SK, Levinson B, et al. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat Genet 2001; 28: 345–9. [Google Scholar]
- Hayflick SJ, Westaway SK, Levinson B, et al. Genetic, clinical and radiographic delineation of Hallervorden-Spatz syndrome. N Engl J Med 2003; 348: 33–40. [Google Scholar]
- Ching KHL, Westaway SK, Levinson B, et al. HARP syndrome is allelic with pantothenate kinaseassociated neurodegeneration. Neurology 2002; 58: 1673–4. [Google Scholar]
- Guillerman P. The eye of the tiger Sign1. Radiology 2000; 217: 895–6. [Google Scholar]
© 2003 médecine/sciences - Inserm / SRMS
Liste des tableaux
Phénotype des neurodégénerescences associées à un déficit en pantothénate kinase.
Liste des figures
|
Figure 1. IRM chez un patient atteint de maladie d’Hallervorden-Spatz: image caractéristique en « œil de tigre » (reproduit de [5]) |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.