")
")
| Issue |
Med Sci (Paris)
Volume 18, Number 10, Octobre 2002
|
|
|---|---|---|
| Page(s) | 975 - 981 | |
| Section | M/S Revues : Articles de Synthèse | |
| DOI | https://doi.org/10.1051/medsci/20021810975 | |
| Published online | 15 October 2002 | |
De nouveaux rôles pour l’ADN topo-isomérase I
New roles for DNA topoisomerase I
Laboratoire de Pharmacologie des agents anti-cancéreux, Institut Bergonié, 229, cours de l’Argonne, 33076 Bordeaux Cedex, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
L’ADN topo-isomérase I eucaryote est une enzyme nucléaire dont le rôle essentiel est de supprimer les contraintes de torsion de l’ADN au cours des processus de réplication et de transcription. La découverte de son activité de protéine kinase, de sa grande similitude avec des enzymes de recombinaison de l’ADN, et de sa capacité de « reconnaître » une grande variété de lésions de l’ADN, a permis d’envisager de nouveaux rôles pour cette enzyme. Nous discuterons en particulier le rôle potentiel de la topo-isomérase I dans la signalisation des dommages de l’ADN aux systèmes de mort cellulaire.
Abstract
DNA topoisomerase I is an nuclear enzyme involved in multiple DNA transactions such as replication, transcription and chromosome condensation. Besides its activity of DNA relaxation, that is essential for the removal of torsional constraints associated with these processes, topoisomerase I may have other functions related to its kinase activity, its similarity with DNA recombinases, and its ability to be trapped by various DNA modifications. In this review, recent findings on the poisoning of topoisomerase I by a broad range of DNA lesions are discussed in the context of a possible role of the enzyme in DNA damage signalling to the cell death machinery.
© 2002 médecine/sciences - Inserm / SRMS
L’ADN topo-isomérase I (topo I) est une enzyme nucléaire ubiquitaire dont la fonction principale est d’assurer le bon déroulement de la condensation et de la décondensation des chromosomes au cours de la division cellulaire. Elle permet de contrôler la topologie de la double hélice d’ADN au cours de ces processus, en supprimant les contraintes de torsion de l’ADN occasionnées par la progression des fourches de réplication ou de transcription [1]. Ce rôle repose principalement sur sa capacité d’introduire transitoirement des coupures simple-brin dans l’ADN [2]. Il existe deux types de topo I qui agissent de façon monomérique mais qui ont une polarité de coupure différente. La topo IA des organismes procaryotes forme une liaison avec l’extrémité 5’ de l’ADN coupé, alors que la topo IB des organismes eucaryotes et des virus se fixe sur l’extrémité 3’ de l’ADN coupé (Figure 1A). Le cycle catalytique de la topo-isomérase I humaine est divisé schématiquement en quatre étapes : une étape de liaison de l’enzyme à l’ADN, une étape de clivage introduisant la coupure, une étape de rotation du brin coupé autour du brin non coupé permettant de diminuer le degré de torsion, et une étape de religation permettant de restaurer la continuité du brin d’ADN (Figure 1B) [1, 3]. Il existe un équilibre entre clivage et religation qui, dans les conditions normales, est déplacé vers la religation. Un grand nombre d’inhibiteurs de topo I (comme la camptothécine) transforment cette enzyme en un poison cellulaire en inhibant l’étape de religation, ce qui se traduit par une « stabilisation » du complexe covalent ADN-enzyme, appelé complexe de clivage [4, 5]. Ces inhibiteurs sont très utilisés dans le traitement d’un grand nombre de cancers et représentent une classe de médicaments anticancéreux en pleine expansion à l’heure actuelle [6, 7]. Il est maintenant admis que ce n’est pas la stabilisation du complexe en elle-même, mais la formation de cassures de l’ADN résultant de cette stabilisation qui est responsable de l’effet cytotoxique de ces médicaments. Le mécanisme de formation de ces lésions implique une collision entre une fourche de réplication (ou de transcription) et le complexe de clivage stabilisé par l’inhibiteur (Figure 1C) et s’appuie sur le fait que l’aphidicoline, un agent bloquant la réplication, est capable de supprimer cette cytotoxicité [5, 8].
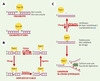 |
Figure 1. Les topo-isomérases eucaryotes et procaryotes. A. La topo IA des organismes procaryotes forme une liaison avec l’extrémité 5’ de l’ADN coupé tandis que la topo IB des organismes eucaryotes et des virus se fixe sur l’extrémité 3’ de l’ADN coupé. B. Les quatre étapes du cycle catalytique de la topo I humaine : liaison de l’enzyme à l’ADN, clivage introduisant la coupure, rotation du brin coupé autour du brin non coupé permettant de diminuer le degré de torsion et religation permettant de restaurer la continuité du brin d’ADN. C. Hypothèse de mécanisme d’endommagement de l’ADN par les poisons de topo I de la famille des camptothécines (CPT). La camptothécine est schématisée par le rectangle bleu. Le pentagone vert schématise le complexe de réplication entrant en collision avec le complexe de clivage stabilisé par la camptothécine. Les flèches pointillées représentent les brins d’ADN nouvellement synthétisés. |
Un certain nombre de résultats inattendus ont permis d’envisager de nouveaux rôles pour la topo I eucaryote, dont certains sont liés spécifiquement à ses activités de coupure ou de religation, et d’autres sont indépendants de son activité catalytique. Le but de cet article est de présenter ces nouveaux rôles, en discutant plus particulièrement des données récentes concernant la reconnaissance par cette enzyme des dommages de l’ADN et la signification biologique de tels résultats.
Topo-isomérase I et transcription
La topo I semble jouer un rôle dans la régulation de la transcription et cela indépendamment de son activité catalytique. Cet éventuel rôle de la topo I repose sur des études d’interaction de l’enzyme avec d’autres facteurs protéiques. La topo I se lie aux protéines du complexe de transcription comme les protéines TBP (TATA-binding protein) et active directement l’étape d’initiation de la transcription en stimulant la formation du complexe TFII-D-TFIIA (→) [9]. L’activité de clivage de l’enzyme n’est pas nécessaire pour observer cet effet puisqu’une topo I catalytiquement inactive, pouvant se lier à l’ADN sans pouvoir le couper, est capable d’induire cette activation. Plus récemment, un système de double-hybride a permis l’identification de protéines nucléaires riches en séquences peptidiques arginine-sérine ou sérine-arginine appelées topors (topo-isomerase I-binding RS proteins) [10]. L’interaction des topors avec la partie-amino-terminale de la topo-isomérase I permettrait le recrutement de l’ARN polymé-rase II directement au niveau des sites de transcription. La même hypothèse a été envisagée pour le recrutement de l’ARN polymérase I, soit par interaction directe de la topo I avec la polymé-rase, soit par interaction indirecte utilisant comme intermédiaire la nucléoline dont la co-localisation avec la topo I au niveau du nucléole a été démontrée [11].
(→) m/s 2000, n°10, p. 1145
En 1996, une étude a également montré que la topo I possédait une activité kinase spécifique pour des protéines riches en domaines arginine-sérine impliquées dans l’épissage d’introns chez certains métazoaires (→) [12]. Cette étude a par ailleurs montré que la phosphorylation de ces facteurs était réduite en présence d’inhibiteurs de topo I. Ces résultats suggèrent donc un rôle de la topo I dans la maturation des ARN prémessagers. Cette hypothèse est renforcée par une étude plus récente ayant montré une régulation de l’activité de la topo I par l’interaction directe de l’enzyme avec un autre facteur d’épissage appelé PSF (pyrimidine tract binding protein associated splicing factor), lui même complexé à des protéines nucléaires se fixant spécifiquement à l’ARN [13]. Cependant, la phosphorylation de ces facteurs d’épissage pouvant être réalisée par d’autres kinases, il reste à déterminer de manière plus spécifique la part de l’activité kinase de la topo I dans le contrôle de l’expression génique.
(→) m/s 1996, n°8-9, p. 1029
Topo-isomérase I et recombinaison de l’ADN
L’équilibre clivage/religation catalysé par la topo I peut être perturbé par de nombreux facteurs, comme les inhibiteurs de l’enzyme ou les altérations de la cible ADN. Ces perturbations peuvent entraîner une stabilisation des complexes de clivage ADN-enzyme. Dans certains cas, cette stabilisation est irréversible et aboutit à la formation de complexes « suicide » possédant une molécule de topo I liée de façon covalente à l’extrémité 3’ de la coupure (Figure 2). Ce type de complexe possède, de par l’activité de religation de l’enzyme, une forte activité de recombinaison in vitro. Toute molécule d’ADN, simple-brin ou double-brin, même non homologue, ayant une extrémité 5’-OH libre, est susceptible d’attaquer la liaison tyrosyl-phosphodiester et de former un produit de recombinaison dite illégitime (Figure 2). Cette propriété de recombinaison in vitro de la topo I du virus de la vaccine [14] a judicieusement été mise à profit pour l’élaboration de systèmes de ligation rapides et performants (TOPO Cloning kits, Invitrogen, Carlsbad, CA, USA).
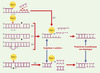 |
Figure 2. Propriétés de recombinaison de la topo I in vitro. Cette activité repose sur la formation de complexes ADNenzyme appelés complexes « suicide » résultant : de la collision du complexe stabilisé par un inhibiteur avec une fourche de réplication (a) ; de la présence d’une interruption de brin soit sur le brin scissile à distance du site de clivage en aval de la coupure (b), soit sur le brin non-scissile à distance du site de coupure (c), ou sous forme d’une boucle de mésappariement en aval du site (d) sur le brin non scissible directement opposé au site (e). L’extrémité 5’-OH de toute molécule d’ADN simple-brin ou double-brin non homologue (traits en pointillés) est capable d’attaquer la liaison covalente ADN-enzyme et d’engendrer un produit de recombinaison illégitime. |
La topo I peut-elle être à l’origine de recombinaisons illégitimes in vivo ? Plusieurs arguments expérimentaux vont dans ce sens (1). Tout d’abord, la similitude de structure et de mécanisme d’action de la topo 1 avec certaines recombinases comme l’intégrase du phage lambda, suggèrent que ces deux enzymes dérivent d’une même ADN-transférase ancestrale [15]. (2) La topo I du virus de la vaccine peut catalyser l’excision du prophage lambda par un mécanisme de recombinaison illégitime [16]. (3) Il existe également une similitude de séquence entre les sites d’intégration de l’ADN des virus SV40 et de l’hépatite B et des sites de coupure pour la topo I eucaryote [17, 18]. Il est donc possible que l’activité de religation de la topo I eucaryote puisse être détournée par certains virus ne contenant pas d’ADN transférase pour permettre leur intégration dans le génome des cellules hôtes. (4) Il existe enfin une corrélation entre l’activité de la topo I et la fréquence des réarrangements chromosomiques chez la levure [19], et (5) une plus grande fréquence d’intégration d’ADN viral dans le génome de levures traitées par un inhibiteur de topo I qui favorise la stabilisation du complexe ADN-enzyme et, par conséquent, les chances de recombinaison illégitime [20]. Il est encore difficile d’appréhender les conséquences de la recombinaison illégitime catalysée par la topo I à l’échelle cellulaire, mais il semblerait que la stabilisation de l’enzyme soit délétère pour les cellules en favorisant une instabilité génomique.
Topo-isomérase I et reconnaissance des dommages de l’ADN
L’ADN génomique est en permanence soumis à des dommages qui peuvent être d’origine endogène (stress oxydatif lié au métabolisme cellulaire, lésions provenant des processus de réparation de l’ADN) ou exogène (agents carcinogènes d’origine industrielle, radiations ionisantes, médicaments anticancéreux,…). Ces lésions sont généralement reconnues par des systèmes de réparation spécifiques dont la tâche est d’assurer l’intégrité du génome. Le fait que les topo I soient des enzymes ubiquitaires, coupant l’ADN à des fréquences élevées, pose le problème de la présence de lésions de l’ADN à proximité de sites de coupure de l’enzyme. Les premières études in vitro ont montré que la présence de dimères de thymine dans de l’ADN plasmidique exposé aux rayonnements ultra-violets était capable d’induire la formation de complexes de clivage ADN-topo I [21]. Au cours des cinq dernières années, des systèmes reposant sur l’utilisation d’oligonucléotides contenant un site unique de clivage pour la topo I (Figure 3) nous ont permis de montrer qu’une grande variété de lésions de l’ADN peuvent perturber l’activité de la topo I in vitro [22]. Nous avons montré que, selon leur nature et leur position par rapport au site de coupure de l’enzyme, certaines lésions induisent une accumulation de complexes de clivage tandis que d’autres inhibent la coupure de l’ADN par la topo I (Figure 4). L’inhibition de coupure est probablement liée à des modifications structurales qui empêchent la liaison de la topo I à son substrat. Ces effets de position reflètent particulièrement bien les spécificités d’interaction qui existent entre la topo I et l’ADN, spécificités qui ont été montrées par les études de cristallographie de l’enzyme en complexe avec son substrat [23, 24]. Le Tableau I regroupe les différentes lésions étudiées jusqu’à présent. À titre d’exemple, la Figure 3B montre que la substitution de la cytosine en position +1 (par rapport au site de coupure) par une cytosine arabinoside (ara-C, un analogue nucléosidique très utilisé en chimiothérapie) induit la formation du complexe de clivage ADN-topo-isomérase I [25]. Pour l’ara-C comme pour beaucoup de lésions étudiées, cet effet est dû à une inhibition réversible de l’étape de religation et s’apparente donc à l’effet que l’on observe en présence d’inhibiteurs de la topo I de la famille des camptothécines (Figure 3). Dans d’autres cas, c’est l’intensification de l’étape de coupure qui est à l’origine de l’augmentation du nombre de complexes de clivage [22]. Enfin, pour des lésions « sévères » de l’ADN, qui induisent une perturbation importante des liaisons hydrogènes à proximité du site de clivage (tels que sites abasiques, interruptions de brin ou adduits carcinogènes volumineux), la topo I est piégée de façon irréversible et il y a formation de complexes suicides particulièrement prompts à créer des recombinaisons (Figure 2). La topo I est donc une enzyme qui est très sensible aux altérations structurales de l’ADN. La validation de tels résultats dans les cellules nécessitait l’utilisation d’un agent génotoxique qui puisse ne produire qu’un seul type de lésion de l’ADN. Notre choix s’est porté sur des agents comme les analogues nucléosidiques tels que l’ara-C ou la gemcitabine qui s’incorporent dans l’ADN génomique de cellules en cours de division. Il fallait également disposer d’une technique permettant de mettre en évidence la formation des complexes ADN-topo I dans les cellules traitées. Grâce au test ICE (immuno complex of enzyme) [26] que nous ne détaillerons pas ici, il a été possible de montrer que le traitement par l’ara-C de cellules leucémiques humaines induisait spécifiquement la formation de complexes ADN-topo I et, qu’effectivement, cette induction était liée à l’incorporation de ce nucléoside dans l’ADN au cours de sa réplication [25]. Ces résultats démontrent que la topo-isomérase I peut être piégée par des modifications structurales de l’ADN au niveau cellulaire. Le fait qu’une lignée leucémique humaine déficiente en topo I, et très résistante à la camptothécine, résiste également à l’ara-C est en accord avec le fait que la séquestration de la topo I pourrait être un des mécanismes d’activité anti-tumorale des analogues nucléosidiques. Ces résultats permettent également d’imaginer un nouveau mode d’inhibition de la topo I par action de toute molécule susceptible de séquestrer cette enzyme en produisant des altérations de la cible ADN à proximité de ses sites de coupure.
 |
Figure 3. Mesure in vitro des activités de coupure et de religation de la topo I. A. Le système utilisé repose sur l’utilisation d’un oligonucléotide dont la séquence provient d’une région particulière de l’ADN ribosomique de Tetrahymena [32]. Cet oligonucléotide de 37-mer contient un site unique de coupure pour l’enzyme (flèche), est radiomarqué à l’extrémité 3’ du brin scissile (*A) et est incubé avec de la topo I purifiée en présence ou non d’inhibiteurs. Il est possible de mesurer la quantité de complexes de clivage formés, qui est directement proportionnelle à la quantité de fragments de 23 bases libérés après la coupure. Ce produit de clivage est distingué du brin non coupé par électrophorèse en gel dénaturant de polyacrylamide. X : positionnement de la lésion par rapport au site de coupure. B. Exemple de séquestration de la topo I liée à la présence d’ara-C. La substitution d’une cytosine en position +1 par l’ara-C (X) provoque une augmentation de la quantité de complexes de clivage ADN-topo I (comparer les lignes 2 et 4 au niveau du produit de coupure de 23 bases). Cette augmentation est proche de celle observée en présence de camptothécine (ligne 3). Ligne 1 : ADN non coupé. |
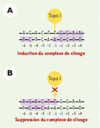 |
Figure 4. Position des lésions de l’ADN et activité de la topo I. A. En règle générale, la présence d’une lésion directement en aval du site de coupure (bases +1 à +4) ainsi qu’à une distance éloignée en amont du site (positions -5 et -6) induit une accumulation du nombre de complexes de clivage ADN-topo I par séquestration de l’enzyme. B. En revanche, quand ces lésions sont présentes directement en amont du site de coupure (positions -1 à -5), l’ADN n’est pas coupé, probablement par un empêchement de la liaison de l’enzyme à son substrat. |
Classification des lésions de l’ADN en fonction de leur effet sur l’ADN topo-isomérase I.
Deux hypothèses sont proposées pour donner une signification biologique au fait que la topo I puisse être sensible à une si grande variété de lésions de l’ADN et soit piégée quand ces lésions sont présentes à proximité d’un site de coupure de l’enzyme.
• Une première hypothèse suggère un rôle de la topo I dans la réparation de l’ADN. Sa séquestration constituerait un « étiquetage » de la lésion qui permettrait un recrutement des protéines de réparation, directement au niveau du site endommagé. L’association de la topo I avec la protéine p53 piégée sur de l’ADN génomique de cellules soumises à un rayonnement ultra-violet est un argument en faveur de cette hypothèse [27]. p53 est un facteur de transcription dont l’expression est stimulée par les dommages de l’ADN et dont l’interaction avec la topo I est connue pour stimuler l’activité catalytique de l’enzyme [28]. p53 exerce un rôle clé dans la réponse cellulaire aux stress génotoxiques en activant un ensemble de gènes jouant un rôle crucial dans l’arrêt du cycle cellulaire nécessaire à la réparation de l’ADN. L’association topo I-p53 permettrait de recruter d’autres facteurs interagissant directement avec p53 tels que GADD45 (growth arrest and DNA damage-inducible gene) qui, en association avec la protéine PCNA (proliferating cell nuclear antigen), est connue pour stimuler la synthèse d’ADN associée au processus de réparation par excision nucléotidique [29]. Dans cette hypothèse, la topo I aurait donc un rôle de protection des cellules vis-à-vis des dommages infligés à l’ADN génomique.
Des résultats récents permettent d’envisager une seconde hypothèse selon laquelle la topo I piégée sur de l’ADN endommagé serait au contraire un obstacle à la réparation des lésions et contribuerait à la mort cellulaire. En effet, si la topo I intervenait dans la réparation, des cellules exprimant fortement cette enzyme devraient être plus résistantes à des agents endommageant l’ADN. Or, il a été montré que des levures surexprimant la topo I étaient en fait plus sensibles aux agents alkylants, aux rayonnements ultra-violets et aux irradiations γ que les levures sauvages correspondantes [30]. L’activité de la topo I est bien responsable de ce phénomène puisque la surerexpression d’une enzyme inactive ne confère pas d’hypersensibilité à ces agents. Dans cette seconde hypothèse, la topo I jouerait un rôle d’indicateur du niveau des lésions de l’ADN. Elle n’exercerait ses effets délétères qu’à partir d’un certain seuil d’altération au-delà duquel la saturation des systèmes de réparation de l’ADN provoquerait une accumulation de lésions et une augmentation du nombre de molécules de topo I piégées sur l’ADN non réparé. Cette séquestration de la topo I servirait à déclencher ou à amplifier un programme de mort cellulaire en participant à la fragmentation de l’ADN, telle une endonucléase au cours du processus d’apoptose. En choisissant cette alternative, la cellule permettrait ainsi d’éviter que n’apparaissent, après la mitose, trop de mutations ou de recombinaisons génomiques. Cette hypothèse est renforcée par une étude récente montrant que la fragmentation de l’ADN de cellules leucémiques traitées par de la staurosporine, un inhibiteur de kinase qui n’est pas connu pour endommager l’ADN, s’accompagne aussi de la formation de complexes de clivage ADN-topo I. L’apoptose induite par la staurosporine est moins importante dans des cellules leucémiques ne contenant pas de topo I que dans les cellules témoins, et s’accompagne d’une moindre fragmentation de l’ADN génomique, suggérant bien une implication de la topo I dans ce type de mort cellulaire [31]. Il reste cependant à déterminer si c’est la séquestration de la topo I qui est un élément déclenchant le processus apoptotique ou si c’est l’ADN endommagé au cours de l’apoptose qui sert de piège pour la topo I, permettant d’amplifier le signal de mort cellulaire.
Conclusions
L’ADN topo-isomérase I a depuis longtemps été considérée comme une enzyme dont l’unique fonction était de relâcher l’ADN pour assurer un bon déroulement des processus de réplication et de transcription. La découverte de son activité de protéine kinase, de sa grande similitude avec des enzymes de recombinaison de l’ADN, et de sa capacité de « reconnaître » une grande variété de lésions de l’ADN, a permis d’envisager de nouveaux rôles pour cette enzyme. La possibilité de piéger la topo I en introduisant des dommages dans l’ADN offre également de nouvelles perspectives, tant au niveau du développement de nouveaux agents anti-cancéreux capables d’inhiber l’enzyme, qu’au niveau de la compréhension des mécanismes par lesquels les complexes ADN-topo I sont réparés au niveau cellulaire.
Remerciements
Je tiens à remercier le Dr A. Jacquemin-Sablon pour ses commentaires à la lecture de ce manuscrit.
Références
- Wang JC. DNA topoisomerases. Annu Rev Biochem 1996; 65 : 635–92. [Google Scholar]
- Champoux JJ. Mechanistic aspects of type-I topoisomerases. In : Wang JC, Cozarelli NR, eds. DNA topology and its biological effects. Cold Spring Harbor, NY : Cold Spring Harbor Laboratory Press, 1990 : 217–42. [Google Scholar]
- Stewart L, Redinbo MR, Qiu X, et al. A model for the mechanism of human topoisomerase I. Science 1998; 279 : 1534–41. [Google Scholar]
- Svejstrup JQ, Christiansen K, Gromova II, et al. New technique for uncoupling the cleavage and religation reactions of eukaryotic topoisomerase I. The mode of action of camptothecin at a specific recognition site. J Mol Biol 1991; 222 : 669–78. [Google Scholar]
- Hsiang YH, Lihou MG, Liu LF. Arrest of replication forks by drug-stabilized topoisomerase I-DNA cleavable complexes as a mechanism of cell killing by camptothecin. Cancer Res 1989; 49 : 5077–82. [Google Scholar]
- Pommier Y, Pourquier P, Urasaki Y, et al. Topoisomerase I inhibitors: selectivity and cellular resistance. Drug Resist Updat 1999; 2 : 307–18. [Google Scholar]
- Bailly C. Topoisomerase I poisons and suppressors as anticancer drugs. Curr Med Chem 2000; 7 : 39–58. [Google Scholar]
- Holm C, Covey JM, Kerrigan D, et al. Differential requirement of DNA replication for the cytotoxicity of DNA topoisomerase I and II inhibitors in Chinese hamster DC3F cells. Cancer Res 1989; 49 : 6365–8. [Google Scholar]
- Shykind BM, Kim J, Stewart L, et al. Topoisomerase I enhances TFIID-TFIIA complex assembly during activation of transcription. Genes Dev 1997; 11 : 397–407. [Google Scholar]
- Haluska P Jr, Saleem A, Rasheed Z, et al. Interaction between human topoisomerase I and a novel RING finger/arginineserine protein. Nucleic Acids Res 1999; 27 : 2538–44. [Google Scholar]
- Bhart AK, Olson MO, Kufe DW, et al. Identification of a nucleolin binding site in human topoisomerase I. J Biol Chem 1996; 271 : 1993–7. [Google Scholar]
- Rossi F, Labourier E, Forne T, et al. Specific phosphorylation of SR proteins by mammalian DNA topoisomerase I. Nature 1996; 381 : 80–2. [Google Scholar]
- Straub T, Grue P, Uhse A, et al. The RNA-splicing factor PSF/p54 controls DNA-topoisomerase I activity by a direct interaction. J Biol Chem 1998; 273 : 26261–4. [Google Scholar]
- Shuman S. Novel approach to molecular cloning and polynucleotide synthesis using vaccinia DNA topoisomerase. J Biol Chem 1994; 269 : 32678–84. [Google Scholar]
- Krogh BO, Shuman S. Catalytic mechanism of DNA topoisomerase IB. Mol Cell 2000; 5 : 1035–41. [Google Scholar]
- Cheng C, Kussie P, Pavletich N, et al. Conservation of structure and mechanism between eukaryotic topoisomerase I and site-specific recombinases. Cell 1998; 92 : 841–50. [Google Scholar]
- Bullock P, Champoux JJ, Botchan, M. Association of crossover points with topoisomerase I cleavage sites: a model for nonhomologous recombination. Science 1985; 230 : 954–8. [Google Scholar]
- Wang HP, Rogler CE. Topoisomerase I-mediated integration of hepadnavirus DNA in vitro. J Virol 1991; 65 : 2381–92. [Google Scholar]
- Zhu J, Schiestl RH. Topoisomerase I involvement in illegitimate recombination in Saccharomyces cerevisiae. Mol Cell Biol 1996; 16 : 1805–12. [Google Scholar]
- Anderson RD, Berger NA. International commission for protection against environmental mutagens and carcinogens. Mutagenicity and carcinogenicity of topoisomerase-interactive agents. Mutat Res 1994; 309 : 109–42. [Google Scholar]
- Lanza A, Tornaletti S, Rodolfo C, et al. Human DNA topoisomerase Imediated cleavages stimulated by ultraviolet light-induced DNA damage. J Biol Chem 1996; 271 : 6978–86. [Google Scholar]
- Pourquier P, Pommier Y. Topoisomerase I-mediated DNA damage. Adv Cancer Res 2001; 80 : 189–216. [Google Scholar]
- Pourquier P, Kohlhagen G, Ueng LM, et al. Topoisomerase I and II activity assays. In : Brown R, Böger-Brown U, eds. Cytotoxic drug resistance mechanisms. Totowa : Humana Press Inc, 1999 : 95–110. [Google Scholar]
- Redinbo MR, Stewart L, Kuhn P, et al. Crystal structures of human topoisomerase I in covalent and noncovalent complexes with DNA. Science 1998; 279 : 1504–13. [Google Scholar]
- Pourquier P, Takebayashi Y, Urasaki Y, et al. Induction of topoisomerase I cleavage complexes by 1-β-D-arabinofuranosylcytosine (ara-C) in vitro and in ara-C-treated cells. Proc Natl Acad Sci USA 2000; 97 : 1885–90. [Google Scholar]
- Subramanian D, Kraut E, Staubus A, et al. Analysis of topoisomerase I/DNA complexes in patients administered topotecan. Cancer Res 1995; 55 : 2097–103. [Google Scholar]
- Mao Y, Okada S, Chang LS, et al. p53 dependence of topoisomerase I recruitment in vivo. Cancer Res 2000; 60 : 4538–43. [Google Scholar]
- Gobert C, Bracco L, Rossi F, et al. Modulation of DNA topoisomerase I activity by p53. Biochemistry 1996; 35 : 5778–86. [Google Scholar]
- Smith ML, Chen IT, Zhan Q, et al. Interaction of the p53-regulated protein Gadd45 with proliferating cell nuclear antigen. Science 1994; 266 : 1376–80. [Google Scholar]
- Nitiss JL, Nitiss KC, Rose A, et al. Overexpression of type I topoisomerases sensitizes yeast cells to DNA damage. J Biol Chem 2001; 276 : 26708–14. [Google Scholar]
- Pourquier P, Kohlhagen G, Urasaki Y, et al. Induction of topoisomerase I cleavage complexes by staurosporine in apoptotic leukemia cells: could topoisomerase I act as an apoptotic endonuclease? Proc Am Assoc Cancer Res 2001; 42 : 303. [Google Scholar]
- Bonven BJ, Gocke E, Westergaard O. A high affinity topoisomerase I binding sequence is clustered at DNAase I hypersensitive sites in Tetrahymena R-chromatin. Cell 1985; 41 : 541–51. [Google Scholar]
Liste des tableaux
Classification des lésions de l’ADN en fonction de leur effet sur l’ADN topo-isomérase I.
Liste des figures
|
Figure 1. Les topo-isomérases eucaryotes et procaryotes. A. La topo IA des organismes procaryotes forme une liaison avec l’extrémité 5’ de l’ADN coupé tandis que la topo IB des organismes eucaryotes et des virus se fixe sur l’extrémité 3’ de l’ADN coupé. B. Les quatre étapes du cycle catalytique de la topo I humaine : liaison de l’enzyme à l’ADN, clivage introduisant la coupure, rotation du brin coupé autour du brin non coupé permettant de diminuer le degré de torsion et religation permettant de restaurer la continuité du brin d’ADN. C. Hypothèse de mécanisme d’endommagement de l’ADN par les poisons de topo I de la famille des camptothécines (CPT). La camptothécine est schématisée par le rectangle bleu. Le pentagone vert schématise le complexe de réplication entrant en collision avec le complexe de clivage stabilisé par la camptothécine. Les flèches pointillées représentent les brins d’ADN nouvellement synthétisés. |
| Dans le texte | |
|
Figure 2. Propriétés de recombinaison de la topo I in vitro. Cette activité repose sur la formation de complexes ADNenzyme appelés complexes « suicide » résultant : de la collision du complexe stabilisé par un inhibiteur avec une fourche de réplication (a) ; de la présence d’une interruption de brin soit sur le brin scissile à distance du site de clivage en aval de la coupure (b), soit sur le brin non-scissile à distance du site de coupure (c), ou sous forme d’une boucle de mésappariement en aval du site (d) sur le brin non scissible directement opposé au site (e). L’extrémité 5’-OH de toute molécule d’ADN simple-brin ou double-brin non homologue (traits en pointillés) est capable d’attaquer la liaison covalente ADN-enzyme et d’engendrer un produit de recombinaison illégitime. |
| Dans le texte | |
|
Figure 3. Mesure in vitro des activités de coupure et de religation de la topo I. A. Le système utilisé repose sur l’utilisation d’un oligonucléotide dont la séquence provient d’une région particulière de l’ADN ribosomique de Tetrahymena [32]. Cet oligonucléotide de 37-mer contient un site unique de coupure pour l’enzyme (flèche), est radiomarqué à l’extrémité 3’ du brin scissile (*A) et est incubé avec de la topo I purifiée en présence ou non d’inhibiteurs. Il est possible de mesurer la quantité de complexes de clivage formés, qui est directement proportionnelle à la quantité de fragments de 23 bases libérés après la coupure. Ce produit de clivage est distingué du brin non coupé par électrophorèse en gel dénaturant de polyacrylamide. X : positionnement de la lésion par rapport au site de coupure. B. Exemple de séquestration de la topo I liée à la présence d’ara-C. La substitution d’une cytosine en position +1 par l’ara-C (X) provoque une augmentation de la quantité de complexes de clivage ADN-topo I (comparer les lignes 2 et 4 au niveau du produit de coupure de 23 bases). Cette augmentation est proche de celle observée en présence de camptothécine (ligne 3). Ligne 1 : ADN non coupé. |
| Dans le texte | |
|
Figure 4. Position des lésions de l’ADN et activité de la topo I. A. En règle générale, la présence d’une lésion directement en aval du site de coupure (bases +1 à +4) ainsi qu’à une distance éloignée en amont du site (positions -5 et -6) induit une accumulation du nombre de complexes de clivage ADN-topo I par séquestration de l’enzyme. B. En revanche, quand ces lésions sont présentes directement en amont du site de coupure (positions -1 à -5), l’ADN n’est pas coupé, probablement par un empêchement de la liaison de l’enzyme à son substrat. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.