")
")
| Issue |
Med Sci (Paris)
Volume 18, Number 10, Octobre 2002
|
|
|---|---|---|
| Page(s) | 925 - 927 | |
| Section | Le Magazine : Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/20021810925 | |
| Published online | 15 octobre 2002 | |
Nouveaux regards sur les mosaïques et chimères humaines
Mosaicism and chimeras
9, rue Basse, 54330 Clerey-sur-Brenon, France
La plupart des êtres humains disposent d’un patrimoine (les 23 chromosomes du spermatozoïde paternel) et d’un matrimoine (les 23 chromosomes de l’ovule maternel), mais certains ont des héritages beaucoup plus complexes. Grâce aux moyens actuels d’investigation du génome, il est possible désormais de déceler d’étranges anomalies de fécondation, qui sont peut-être plus nombreuses qu’on ne l’imaginait jusqu’alors. Il faut donc désormais y penser et les rechercher dans certains processus pathologiques.
Hermaphrodites vrais1 et autres jumeaux fusionnés
Dès les années 1970, l’étude du caryotype démontra que certains sujets présentant, comme seule manifestation clinique, une ambiguïté sexuelle, étaient en réalité des êtres doubles, produits de la fusion précoce de deux zygotes. Cette ambiguïté peut du reste passer inaperçue.
Ainsi, elle n’avait pas été notée chez ce garçon, enregistré comme tel à la naissance. Lorsque vers 14 ans, en 1987, il développa une gynécomastie, on découvrit qu’il était porteur d’un caryotype 46,XX/46,XY, avec un ovaire « cryptorchide » responsable de ce début de puberté féminine. Il fallut alors se résoudre à l’ablation de la seule glande sexuelle fonctionnelle et recourir à un traitement hormonal mâle substitutif, car cet enfant, s’identifiant comme un garçon, refusait farouchement de se voir transformé en fille [1]. À l’époque, la connaissance du génome humain était insuffisante pour permettre de retrouver l’origine du matériel génétique de chacun des deux zygotes qui avaient fusionné pour lui donner naissance.
Devant de tels sujets, tout au plus pouvaiton faire l’hypothèse de la fusion de deux œufs s’il existait à la fois un double héritage paternel et maternel ; s’il n’existait qu’un seul héritage maternel pour deux héritages paternels, c’est le second globule polaire qui était supposé fournir la part féminine du deuxième zygote (Figure 1). Plus récemment, par l’analyse moléculaire des deux populations cellulaires, un auteur anglais a pu démontrer, chez un autre hermaphrodite vrai, la double origine zygotique. L’analyse de différents tissus démontra en outre que la répartition des cellules masculines et des cellules féminines n’était pas homogène [2].
 |
Figure 1. Mosaïques et chimères. A. Jumeaux monozygotes. B. Fusion de deux zygotes pour former un seul individu, éventuellement hermaphrodite si les deux spermatozoïdes ont un gonosome différent (d’où un seul œuf avec 2 garnitures gonosomiques XX et XY). On trouve un double héritage paternel et maternel. C. Fusion de deux zygotes avec un seul héritage maternel, par utilisation du deuxième globule polaire. D. Jumeaux dizygotes avec chimère sanguine par anastomose placentaire. n représente le nombre haploïde de chromosomes. |
Ce même auteur a observé une autre anomalie de fécondation : un enfant partiellement parthénogénétique. Dans son sang, et dans certains tissus, il existait des cellules possédant uniquement un héritage maternel. S’il est impossible d’obtenir des enfants par parthénogenèse, ne serait-ce qu’en raison des gènes soumis à empreinte, cette parthénogenèse, parfaitement viable, peut s’expliquer par la présence d’un zygote normal, fusionné avec un ovocyte dont le matériel génétique se serait dupliqué ou qui aurait conservé le deuxième globule polaire [3].
Puisque de telles fusions existent, il est probable qu’elles se produisent aussi avec des zygotes de sexe identique XX/XX ou XY/XY. Mais, dans ces cas, il s’agit de sujets dépourvus de toute particularité. Seule, la présence, lors d’une étude des groupes sanguins, d’une double population d’érythrocytes permet les déceler. Ils peuvent donc passer inaperçus et leur nombre est sans doute sous-estimé. Cette ignorance porte peut-être à conséquence. Pour ne donner qu’un seul exemple, la réponse aux médicaments, qui dépend en partie des facteurs génétiques, peut s’en trouver modifiée (→).
(→) m/s 2002, n°4, p. 389
Chimères sanguines
Une double population d’érythrocytes peut être révélatrice d’un autre phénomène. En effet, les jumeaux dizygotes peuvent, au cours du développement embryonnaire, échanger des cellules par anastomoses placentaires (Figure 1). Si les cellules souches se fixent précocement dans la moelle osseuse de l’autre jumeau, elles ne seront pas rejetées ultérieurement par le système immunitaire et constitueront une chimère sanguine qui pourra perdurer pendant des années, voire toute la vie. On estime que ce phénomène se produit chez 8 % des jumeaux dizygotes [4]. Pour certaines personnes, la surprise est d’autant plus grande qu’elles ignoraient avoir eu un jumeau, aperçu parfois en échographie fœtale au début de la grossesse, et disparu par la suite [5].
Le pourcentage des cellules provenant de l’autre jumeau est variable, mais il arrive que les cellules étrangères colonisent complètement la moelle pour constituer une chimère totale. Ainsi, chez une fillette trisomique 21, les caryotypes établis à partir du sang circulant montraient exclusivement un caryotype 46,XY, alors qu’aucune cellule 47,XX,+21 n’était observée chez son frère jumeau [6]. Sans doute les cellules normales possèdent-elles un avantage sélectif, mais ces cas sont si rares qu’il est difficile d’en faire la preuve. Toutefois, la pratique de la fécondation in vitro a considérablement augmenté le nombre des grossesses multiples, et par conséquent le nombre des sujets porteurs de chimère sanguine.
Mais point n’est besoin d’avoir un frère jumeau pour receler une chimère. Entre la mère et le fœtus, des échanges bilatéraux existent au cours de la grossesse. On sait que certaines femmes conservent dans leur sang pendant des années des cellules XY reçues d’un fœtus mâle. Inversement, il a été démontré récemment que certains sujets conservent à l’âge adulte un microchimérisme d’origine maternelle [7]. Enfin, des transfusions répétées peuvent aussi entraîner chez certains sujets des microchimérismes. Ceux-ci peuvent-ils avoir des conséquences pathologiques ? Peuvent-ils par exemple expliquer l’excès de maladies auto-immunes observé chez les femmes par rapport aux hommes ? Certaines maladies, comme la cirrhose biliaire primitive ou le syndrome de Sjögrenont des points communs avec la réaction du rejet contre l’hôte, observée après transplantation de moelle osseuse. Des recherches de microchimérisme sont actuellement entreprises dans des maladies de ce type. Ils ont été retrouvés dans des éruptions polymorphes de la grossesse [8]. Des microchimérismes d’origine maternelle sont présents plus fréquemment chez des enfants atteints de dermatomyosite que chez leurs frères et sœurs pris comme témoins [9].
Mosaïques tissulaires
Le piège de la tétrasomie 12p est bien connu des cytogénéticiens : cette grave aberration chromosomique ne peut être mise en évidence ni dans le liquide amniotique ni dans les prélèvements de sang (où les caryotypes sont habituellement normaux), alors qu’elle existe dans tous les autres tissus de l’organisme des sujets atteints [10]. Il s’agit d’une anomalie exceptionnelle. Mais des travaux récents ont aussi montré que certains patients atteints de maladie d’Alzheimer étaient porteurs de trisomie 21 en mosaïque, surtout dans des cas familiaux, avec mutation des gènes codant pour les présénilines 1 et 2 [11]. Ces mosaïques pourraient être localisées dans certains tissus. Par exemple, dans le syndrome Proteus, une anomalie en mosaïque expliquerait l’extrême variabilité du phénotype, la survenue sporadique, et la discordance observée chez des couples de jumeaux monozygotes. Des recherches dans ce sens sont en cours aux États-Unis (à l’Institut de Recherche de Bethesda, Maryland), dans cette maladie congénitale qui comporte des hyperplasies monstrueuses du tissu conjonctif, avec malformations vasculaires et anomalies linéaires verruqueuses de l’épiderme [12]. Dans le même ordre d’idée, les lignes de Blaschko2, qui révèlent une hétérogénéité des cellules de l’ectoderme, pourraient être plus systématiquement recherchées car - indépendamment de l’incontinentia pigmenti - elles sont révélatrices de mosaïques (Figure 2). Des études non encore publiées viennent justement de montrer que 10 % des enfants autistes auraient des lignes de Blaschko lorsqu’on les observe sous lumière ultraviolette [13].
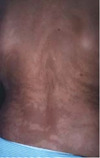 |
Figure 2. Lignes de Blaschko dans la région dorsale d’une femme atteinte d’incontinentia pigmenti. |
Conclusions
On le voit, les « erreurs » de fécondation se révèlent beaucoup plus complexes qu’on ne le supposait il y a une vingtaine d’années, quand on ne disposait que du caryotype et des groupes sanguins pour les étudier. Certains sujets, normaux ou pathologiques, possèdent indiscutablement plusieurs populations cellulaires Celles-ci peuvent être réparties de façon homogène, ou localisées dans certains tissus. Elle peuvent être normales et révéler simplement une double origine parentale ; elles peuvent être pathologiques, avec une lignée porteuse soit d’une anomalie chromosomique, soit d’une mutation génique. Certes, jusqu’à présent, ces recherches ont été peu fructueuses en pathologie humaine car elles n’ont guère aidé à comprendre l’étiologie de maladies, supposées génétiques, pour lesquelles la génétique moléculaire est restée impuissante. Mais elles démontrent que certains êtres sont plus complexes qu’il n’y paraît et proposent de nouveaux champs d’investigation pour les explorer.
L’hermaphrodite vrai possède des gonades ou du tissu gonadique masculin et féminin (ovaire, testicule ou ovotes-tis), contrairement aux pseudo-hermaphrodites qui ne possèdent qu’un type de gonade.
Lignes imaginaires séparant, à la surface de la peau, les territoires en rapport avec des racines nerveuses distinctes.
Références
- Gilgenkrantz S. True hermaphroditism and double fertilization. J Genet Hum 1987; 35 : 105–18. [Google Scholar]
- Strain L, Dean JC, Hamilton MP, Bronthron DT. A true hermaphrodite chimaera resulting from embryo amalgamation after in vitro fertilization. N Engl J Med 1998; 338 : 166–9. [Google Scholar]
- Strain L, Warner JP, Johnston T, Bronthron DT. A human parthenogenetic chimaera. Nat Genet 1995; 11 : 164–9. [Google Scholar]
- Van Dijk BA, Boomsma DL, de Man AJ. Blood group chimerism in human multiple births is not rare. Am J Med Genet 1996; 61 : 264–8. [Google Scholar]
- Boklage CE. Survival probability of human conceptions from fertilization to term. Int J Fertil 1990; 35 : 79–94. [Google Scholar]
- Gilgenkrantz S, Janot C. Monozygotic twins discordant for trisomy 21 or chimeric dizygotic twins ? Am J Med Genet 1983; 15 : 159–60. [Google Scholar]
- Nelson JL. Microchimerism : incidental byproduct of pregnancy or active participant in human health ? Trends Mol Med 2002; 8 : 109–13. [Google Scholar]
- Aractingi S, Berkane N, Bertheau P, et al. Fetal DNA in skin of polymorphic eruptions of pregnancy. Lancet 1999; 352 : 1898–901. [Google Scholar]
- Artlett CM, Ramos R, Jiminez SA, Patterson K, Miller FW, Rider LG. Chimeric cells of maternal origin in juvenile idiopathic inflammatory myopathies. Lancet 2000; 356 : 2155–6. [Google Scholar]
- Mathieu M, Piussan C, Thepot F, et al. Collaborative study of mosaic tetrasomy 12p or Pallister-Killian syndrome (nineteen fetuses or children). Ann Genet 1997; 40 : 45–54. [Google Scholar]
- Geller LN, Potter H. Chromosome missegregation and trisomy 21 mosaicism in Alzheimer’s disease. Neurobiol Dis 1999; 6 : 167–79. [Google Scholar]
- Biesecker LG, Peters KF, Darling TN, et al. Clinical differentiation between Proteus syndrome and hemihyperplasia : description of a disctinct form of hemihyperplasia. Am J Med Genet 1998; 79 : 311–8. [Google Scholar]
- Pearson H. Dual identities. Nature 2002; 417 : 10–1. [Google Scholar]
© 2002 médecine/sciences - Inserm / SRMS
Liste des figures
|
Figure 1. Mosaïques et chimères. A. Jumeaux monozygotes. B. Fusion de deux zygotes pour former un seul individu, éventuellement hermaphrodite si les deux spermatozoïdes ont un gonosome différent (d’où un seul œuf avec 2 garnitures gonosomiques XX et XY). On trouve un double héritage paternel et maternel. C. Fusion de deux zygotes avec un seul héritage maternel, par utilisation du deuxième globule polaire. D. Jumeaux dizygotes avec chimère sanguine par anastomose placentaire. n représente le nombre haploïde de chromosomes. |
| Dans le texte | |
|
Figure 2. Lignes de Blaschko dans la région dorsale d’une femme atteinte d’incontinentia pigmenti. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.