")
")
| Issue |
Med Sci (Paris)
Volume 41, Number 11, Novembre 2025
|
|
|---|---|---|
| Page(s) | 842 - 844 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/2025212 | |
| Published online | 12 December 2025 | |
Maladie de Huntington : une accélération du vieillissement neuronal par érosion épigénétique
Huntington’s disease: epigenetic erosion resulting in accelerated neuronal aging
Laboratoire de neurosciences cognitives et adaptatives, CNRS UMR 7364, Université de Strasbourg, Strasbourg, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
La maladie de Huntington : une maladie neurodégénérative héréditaire affectant sélectivement certains neurones
La maladie de Huntington est une maladie héréditaire rare, de transmission autosomique dominante. Elle est en général diagnostiquée à l’âge adulte, vers l’âge de 35 ou 40 ans. Néanmoins, des formes plus rares, plus précoces et plus sévères existent également. La maladie se manifeste par une triade de symptômes moteurs, cognitifs et psychiatriques. Si la présence de mouvements choréiques involontaires est particulièrement évocatrice de cette maladie, l’atteinte des fonctions exécutives et la présence de troubles psychiques altèrent également la qualité de vie des patients. La maladie de Huntington reste aujourd’hui incurable : elle cause la mort après 10 à 15 années d’évolution des symptômes. La maladie est due à une mutation unique : une augmentation du nombre de triplets CAG consécutifs dans l’exon 1 du gène Huntingtine (HTT) [1]. Chez les individus sains, ce nombre est variable, mais n’excède pas 35 répétitions, à la différence des personnes qui seront atteintes de la maladie, chez lesquelles il excède cette valeur. Le codon CAG étant traduit en glutamine, la mutation conduit à la production d’une protéine HTT comportant une expansion du motif polyglutamine (polyQ) qui confère à cette protéine des propriétés nouvelles, dont la propension à former des agrégats toxiques pour les neurones [2]. Bien que la protéine HTT soit ubiquitaire, sa mutation affecte préférentiellement certains neurones, et plus particulièrement les neurones épineux moyens du striatum. La compréhension du mécanisme impliqué dans la sélectivité de cette vulnérabilité neuronale représente un enjeu majeur, car le développement de thérapies efficaces pourrait en découler [3].
Les études effectuées ces dix dernières années mettent en avant un rôle déterminant de l’instabilité de la mutation dans ce mécanisme [4]. En effet, l’expansion des triplets CAG dans le gène HTT est dynamique : leur nombre tend à augmenter dans les cellules germinales au fil des générations, ainsi que dans certaines cellules somatiques avec l’âge [5]. Si l’instabilité de la mutation dans les cellules germinales est à l’origine du phénomène d’anticipation (la maladie devient de plus en plus sévère et précoce au fil des générations), l’instabilité dans les cellules somatiques, qui entraîne la production de protéines HTT-polyQ dont la toxicité s’accroît, est un élément clé du mécanisme pathogénique. Cette conclusion s’appuie sur plusieurs arguments. Premièrement, l’instabilité de la mutation est forte dans les neurones épineux moyens [6]. Deuxièmement, les résultats d’études d’association pangénomiques montrent que les gènes modificateurs de la maladie sont principalement des gènes de réparation de l’ADN capables de moduler l’instabilité de la mutation dans les cellules somatiques [7]. Troisièmement, les résultats d’études de profilage moléculaire effectuées à partir de tissu cérébral provenant de patients ou de souris modèles de la maladie indiquent que l’instabilité de la mutation est nécessaire au processus pathogénique [8, 9]. Plus précisément, ils montrent que cette instabilité conduit progressivement à l’apparition d’une très longue suite de triplets CAG dans le gène HTT muté des neurones épineux moyens. Ces très longues expansions sont à l’origine d’une dérégulation de l’expression génique, entraînant une érosion de l’identité de ces neurones : on constate en effet une diminution de l’expression de gènes marqueurs de leur identité, accompagnée de la réexpression de gènes exprimés pendant le développement, lors du processus de différenciation cellulaire. La réactivation de ces gènes du développement, qui sont normalement réprimés dans les neurones épineux moyens matures, serait particulièrement toxique pour ces neurones et contribuerait à leur dégénérescence [8]. Le mécanisme qui sous-tend la perte d’identité des neurones épineux moyens dans la maladie de Huntington n’est pas encore élucidé. Notre étude apporte néanmoins des éléments de réponse en proposant un rôle critique des régulations épigénétiques [10].
L’identité cellulaire et l’information épigénétique : des processus étroitement liés
En quoi consistent ces régulations épigénétiques ? Si l’information génétique (le génome), décryptée grâce au code génétique, est identique dans toutes les cellules d’un organisme, l’information épigénétique est, quant à elle, spécifique de chaque type cellulaire. Un neurone, un myocyte, ou un hépatocyte assurent des fonctions distinctes qui nécessitent l’activation de programmes d’expression génique différents. Les mécanismes épigénétiques permettent l’activation d’un programme génique adapté à chaque fonction cellulaire, en exerçant un contrôle local du degré de compaction de la chromatine, et donc de l’accessibilité des gènes à la machinerie transcriptionnelle. Méthylation de l’ADN et modifications post-traductionnelles des histones, qui sont des composants majeurs de la chromatine, constituent deux mécanismes épigénétiques clés. Les propriétés physico-chimiques des histones sont ainsi déterminées par la présence ou l’absence de modifications chimiques telles que l’acétylation ou la méthylation : l’acétylation des histones, par exemple l’acétylation de l’histone H3 sur le résidu lysine situé en position 27 (H3K27ac), promeut un état relâché de la chromatine (euchromatine) qui facilite la transcription des gènes ; au contraire, la triméthylation de H3K27 (H3K27me3) favorise un état compacté de la chromatine (hétérochromatine), qui est associé à une répression transcriptionnelle. Les régulations épigénétiques sont notamment cruciales lors du développement d’un organisme multicellulaire, en permettant aux cellules de cet organisme de se différencier en sous-types distincts, et ainsi d’acquérir une identité propre leur permettant d’assurer des fonctions spécifiques [11]. Les régulations épigénétiques jouent également un rôle critique lors du vieillissement cellulaire. Selon la théorie de l’information du vieillissement cellulaire, que corroborent les résultats d’une étude américaine récente, le vieillissement cellulaire serait même causé par une érosion de l’information épigénétique, entraînant une perte d’identité et de fonction [12]. Ainsi, dans une cellule vieillissante, la quantité de H3K27ac diminue dans la chromatine associée aux gènes d’identité de cette cellule, alors que celle de H3K27me3 diminue dans la chromatine associée aux gènes développementaux, normalement inactifs, ce qui entraîne une baisse de l’expression des gènes d’identité et une hausse de l’expression de gènes développementaux. Soulignons que ces avancées conceptuelles ont été possibles grâce à l’utilisation de techniques de séquençage à haut débit (ChIP-sequencing, CUT&Tag, etc.) qui permettent d’analyser les modifications post-traductionnelles des histones dans le génome entier et, par une analyse bioinformatique, d’établir une cartographie précise des modifications épigénétiques dans les cellules [13, 14].
Une accélération de l’érosion épigénétique dans les neurones vulnérables à la mutation responsable de la maladie de Huntington
Afin d’identifier des signatures épigénétiques propres à la maladie de Huntington, nous avons utilisé ces approches de séquençage à haut-débit pour effectuer une analyse temporelle de l’évolution de la quantité de H3K27ac et de H3K27me3 dans les neurones épineux moyens de souris modèles de la maladie. Nous avons ainsi mis en évidence une accélération du mécanisme d’érosion épigénétique dans ces neurones, entraînant leur vieillissement prématuré [10] (Figure 1). D’un point de vue mécanistique, le vieillissement accéléré des neurones du striatum de ces souris implique le complexe répresseur polycomb 1 (PRC1) et une marque épigénétique contrôlée par ce complexe : l’ubiquitinylation de H2AK119 (H2AK119ub), une modification post-traductionnelle de l’histone H2A favorisant la propagation de l’hétérochromatine. Plus précisément, la dérépression des gènes développementaux dans les neurones épineux moyens des souris modèles de la maladie est corrélée non seulement avec une diminution de la quantité de H3K27me3, mais aussi avec une déplétion en H2AK119ub. Le mécanisme en cause n’est pas encore élucidé, mais met vraisemblablement en jeu une permutation des paralogues de la protéine CBX (chromobox), une sous-unité du complexe PRC1 impliquée dans la compaction de la chromatine. En effet, dans les neurones épineux moyens de la maladie de Huntington, l’expression de paralogues de CBX associés aux étapes précoces de la différenciation neurale est favorisée, au détriment de ceux exprimés dans les neurones matures. Une fonction clé de PRC1 est de maintenir l’intégrité du génome en coordonnant la régulation de l’expression génique et les mécanismes de réparation de l’ADN. Existet-il un lien entre la dérégulation de PRC1 et l’instabilité de l’expansion CAG dans ces neurones ? Notre étude ne permet pas de répondre à la question, mais celle-ci mérite d’être posée. En effet, l’instabilité de l’expansion des triplets CAG met en jeu des cassures de l’ADN et des mécanismes de réparation de l’ADN spécifiques. De plus, il semble qu’un processus essentiel à l’érosion épigénétique soit l’accumulation de cassures de l’ADN et leur réparation imparfaite [12]. Quoi qu’il en soit, l’identification des mécanismes épigénétiques à l’origine de la perte d’identité des neurones épineux moyens dans la maladie de Huntington pourrait fournir une piste thérapeutique nouvelle, qui consisterait à freiner le processus d’érosion épigénétique afin de « rajeunir » ces neurones.
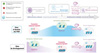 |
Figure 1 Accélération de l’érosion épigénétique dans les neurones vulnérables à la mutation responsable de la maladie de Huntington. Schéma représentant l’état de la chromatine de cellules neuronales au cours de la différenciation et du vieillissement cellulaires. Les neurones épineux moyens (medium spiny neurons, MSN) du striatum, qui sont vulnérables à la mutation responsable de la maladie de Huntington, subissent une érosion épigénétique accélérée dans cette maladie. Tout comme les neurones vieillissants, ils présentent une réduction des marques d’activation transcriptionnelle (e.g., H3K27ac) dans les gènes d’identité neuronale, et au contraire, une augmentation de ces marques dans les gènes développementaux, ainsi que des changements opposés pour les marques associées à la répression transcriptionnelle (e.g., H3K27me3 et H2AK119ub). Cette érosion épigénétique entraîne une baisse d’expression des gènes d’identité neuronale et une réexpression de gènes développementaux, à l’origine d’une perte d’identité et d’un vieillissement prématuré de ces neurones. |
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Références
- Gusella JF, Lee JM, MacDonald ME. Huntington’s disease: nearly four decades of human molecular genetics. Hum Mol Genet 2021 ; 30 : R254-R63. [Google Scholar]
- Bates GP, Dorsey R, Gusella JF, et al. Huntington disease. Nat Rev Dis Primers 2015 ; 1 : 15005. [Google Scholar]
- Tabrizi SJ, Flower MD, Ross CA, Wild EJ. Huntington disease: new insights into molecular pathogenesis and therapeutic opportunities. Nat Rev Neurol 2020 ; 16 : 529–46. [Google Scholar]
- Cattaneo E, Scalzo D, Zobel M, et al. When repetita no-longer iuvant: somatic instability of the CAG triplet in Huntington’s disease. Nucleic Acids Res 2025 ; 53 : gkae1204.. [Google Scholar]
- Pearson CE, Nichol Edamura K, Cleary JD. Repeat instability: mechanisms of dynamic mutations. Nat Rev Genet 2005 ; 6 : 729–42. [CrossRef] [PubMed] [Google Scholar]
- Mouro Pinto R, Arning L, Giordano JV, et al. Patterns of CAG repeat instability in the central nervous system and periphery in Huntington’s disease and in spinocerebellar ataxia type 1. Hum Mol Genet 2020 ; 29 : 2551–67. [Google Scholar]
- Genetic modifiers of Huntington’s disease (GeM-HD) consortium. CAG repeat, not polyglutamine length, determines timing of Huntington’s disease onset. Cell 2019 ; 178 : 887-900 e14. [CrossRef] [PubMed] [Google Scholar]
- Handsaker RE, Kashin S, Reed NM, et al. Long somatic DNA-repeat expansion drives neurodegeneration in Huntington’s disease. Cell 2025 ; 188 : 623-39 e19. [CrossRef] [PubMed] [Google Scholar]
- Wang N, Zhang S, Langfelder P, et al. Distinct mismatch-repair complex genes set neuronal CAG-repeat expansion rate to drive selective pathogenesis in HD mice. Cell 2025 ; 188 : 1524-44 e22. [Google Scholar]
- Brulé B, Alcala-Vida R, Penaud N, et al. Accelerated epigenetic aging in Huntington’s disease involves polycomb repressive complex 1. Nat Commun 2025 ; 16 : 1550. [Google Scholar]
- Atlasi Y, Stunnenberg HG. The interplay of epigenetic marks during stem cell differentiation and development. Nat Rev Genet 2017 ; 18 : 643–58. [CrossRef] [PubMed] [Google Scholar]
- Yang JH, Hayano M, Griffin PT, et al. Loss of epigenetic information as a cause of mammalian aging. Cell 2023 ; 186 : 305-26 e27. [CrossRef] [PubMed] [Google Scholar]
- Bonev B, Castelo-Branco G, Chen F, et al. Opportunities and challenges of single-cell and spatially resolved genomics methods for neuroscience discovery. Nat Neurosci 2024 ; 27 : 2292–309. [Google Scholar]
- Stricker SH, Koferle A, Beck S. From profiles to function in epigenomics. Nat Rev Genet 2017 ; 18 : 51–66. [Google Scholar]
© 2025 médecine/sciences – Inserm
 Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Liste des figures
|
Figure 1 Accélération de l’érosion épigénétique dans les neurones vulnérables à la mutation responsable de la maladie de Huntington. Schéma représentant l’état de la chromatine de cellules neuronales au cours de la différenciation et du vieillissement cellulaires. Les neurones épineux moyens (medium spiny neurons, MSN) du striatum, qui sont vulnérables à la mutation responsable de la maladie de Huntington, subissent une érosion épigénétique accélérée dans cette maladie. Tout comme les neurones vieillissants, ils présentent une réduction des marques d’activation transcriptionnelle (e.g., H3K27ac) dans les gènes d’identité neuronale, et au contraire, une augmentation de ces marques dans les gènes développementaux, ainsi que des changements opposés pour les marques associées à la répression transcriptionnelle (e.g., H3K27me3 et H2AK119ub). Cette érosion épigénétique entraîne une baisse d’expression des gènes d’identité neuronale et une réexpression de gènes développementaux, à l’origine d’une perte d’identité et d’un vieillissement prématuré de ces neurones. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.