")
")
| Issue |
Med Sci (Paris)
Volume 37, Novembre 2021
Les Cahiers de Myologie
|
|
|---|---|---|
| Page(s) | 25 - 29 | |
| Section | Mise au point | |
| DOI | https://doi.org/10.1051/medsci/2021187 | |
| Published online | 8 décembre 2021 | |
Le registre national SMA France : des résultats déjà encourageants
The SMA France national registry: already encouraging results
1
URC APHP Paris-Saclay, France
2
Centre de Référence des Maladies Neuromusculaires, Filnemus, Université Paris Saclay, Garches, France
3
European Reference Center Network (Euro NMD ERN)
4
Institut de Myologie, Paris, France
* Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
L’amyotrophie spinale proximale liée au gène SMN1 (SMA) est une maladie neuromusculaire invalidante dont l’histoire naturelle a été sensiblement modifiée ces dernières années du fait de l’apparition de thérapies innovantes. Le registre SMA France a été mis en place en 2020 afin de mieux connaître la pathologie et répondre au besoin de données en vie réelle induit par l’arrivée de ces nouveaux médicaments. Le but est aussi d’essayer d’identifier les meilleures stratégies thérapeutiques et d’améliorer in fine la prise en charge de ces patients. Ce registre a le statut d’entrepôt de données de santé et collecte des informations rétrospectives et prospectives de patients SMA de tous types, génétiquement confirmés, traités ou non par thérapies innovantes, et suivis dans les centres du réseau FILNEMUS. La population-cible est estimée à 1 000 patients, dont la moitié d’âge pédiatrique. Au 1er octobre 2021, 666 patients ont été inclus dans la base (357 enfants et 309 adultes) par 44 des 51 centres spécialisés du réseau neuromusculaire FILNEMUS ayant accepté de participer. Parmi ces patients, 150 étaient de type 1 (22 %), 278 (42 %) de type 2, 232 (35 %) de type 3, et 4 de type 4 (1 %).
Abstract
Spinal muscular atrophy is a debilitating neuromuscular disease due to the deletion of the SMN1 gene (SMA). The emergence of innovative targeted therapies changed the natural history of this condition. The French registry of SMA (Registre SMA France) was launched in 2020 to obtain a better knowledge of the pathology. The goal of the register was also to meet the need for real-life data regarding the arrival of these innovative therapies in order to identify the best therapeutic strategies and to improve patient care. The registry collects retrospective and prospective data of all genetically confirmed SMA, treated or not treated, in reference centers belonging to the national neuromuscular network (FILNEMUS). The estimated enrollment is 1,000 patients (50% children). On October 1st, 666 patients have been enrolled (357 children and 309 adults) by 44 out of 51 open centers of the national network (FILNEMUS) with: 150 type 1 (22%); 278 type 2 (42%), 232 type 3 (35%) and 4 type 4 (1%) respectively.
ont contribué de manière identique
© 2021 médecine/sciences – Inserm

© Valérie Allamand
Introduction
L’amyotrophie spinale proximale liée au gène SMN1 (aussi connue sous le terme de SMA, pour spinal muscular atrophy) est une maladie neuromusculaire sévère caractérisée par une atrophie et une faiblesse musculaire progressive, conduisant à une paralysie irréversible. L’histoire naturelle de la maladie a été modifiée ces dernières années suite à l’apparition de thérapies innovantes ciblées. Cette révolution thérapeutique est à l’origine d’enjeux importants au niveau clinique, économique et éthique.
Le registre français des amyotrophies spinales – ou registre SMA France – a été mis en place dès janvier 2020 en vue d’améliorer les connaissances générales sur la maladie tout en essayant de répondre au besoin de données en vie réelle des patients atteints de SMA pris en charge en France.
SMA
Physiopathologie
La SMA est une maladie génétique invalidante causée par des délétions récessives ou des mutations ponctuelles du gène SMN1 (situé sur le chromosome 5q) entraînant une dégénérescence des motoneurones alpha de la corne antérieure de la moelle épinière et conduisant à une atrophie qui peut aussi affecter la fonction respiratoire dans les formes les plus graves. La sévérité de la pathologie est très variable et montre un continuum allant des formes les plus sévères (à début prénatal) aux formes les plus bénignes débutant à l’âge adulte.
Les différences phénotypiques s’expliquent, pour partie, par la présence d’un deuxième gène, SMN2 (homologue centromérique de SMN1), qui fournit une quantité résiduelle variable de protéine SMN fonctionnelle. Cette suppléance permet d’expliquer l’existence de phénotypes moins sévères.
La classification actuelle des patients atteints de SMA comprend quatre types, de 1 (forme la plus sévère) à 4, en fonction de l’âge d’apparition des premiers symptômes et les capacités motrices fonctionnelles maximales acquises. Des formes intermédiaires au pronostic différent ont été décrites augmentant le spectre clinique de la maladie. Ces différents types et sous-types sont décrits dans le Tableau I [3].
Classification des différents types et sous-types de SMA [3].
Prévalence
La prévalence de la SMA est variable selon les pays, avec une prévalence moyenne d’environ 20 pour 100 000 naissances vivantes en Europe. Elle représente la cause monogénique la plus fréquente de mortalité infantile [1,2]. L’incidence estimée en Europe est de 2,6 pour 100 000 se répartissant selon les types de SMA comme suit : 0,26/100 000 pour les patients de type 1 ; 1,23 pour le type 2 ; 1,1 pour le type 3 et 0,32 pour le type 4. La proportion des types à la naissance est également détaillée dans le Tableau I [3].
Diagnostic
En pratique courante, le diagnostic de l’amyotrophie spinale est très exceptionnellement posé en l’absence de symptômes cliniques. Ceux-ci sont généralement très évocateurs, même si l’on note, dans un nombre non négligeable de cas, une certaine errance diagnostique. Le diagnostic de la SMA repose sur les antécédents familiaux éventuels et sur la suspicion clinique à l’examen. Le diagnostic est rapidement confirmé par un test génétique spécifique (recherche des mutations du gène SMN1 en biologie moléculaire) [2].
Thérapies innovantes
La prise en charge d’un patient atteint de SMA est symptomatique et nécessite une approche pluridisciplinaire médico-chirurgicale, éducationnelle, psychologique et sociale. Cette prise en charge comprend notamment la kinésithérapie, l’ergothérapie, une assistance respiratoire (ventilation invasive ou non invasive) et/ou nutritionnelle (gastrostomie) selon les cas. Au cours des dernières années, plusieurs thérapies spécifiques ciblant la SMA ont été développées par l’industrie pharmaceutique et leur utilisation a conduit à une évolution des recommandations internationales en matière de standards de soins. Initialement cantonnés à l’accompagnement et aux soins palliatifs, surtout dans les formes les plus sévères, les soins sont désormais plus pro-actifs. Trois thérapies innovantes sont aujourd’hui disponibles en France.
Spinraza® (nusinersen)
Le Spinraza® est le premier traitement à avoir obtenu une autorisation de mise sur le marché (AMM) en Europe en 2017. Son principe actif, le nusinersen, est un oligonucléotide antisens qui augmente la production de protéine SMN fonctionnelle en agissant sur l’épissage du gène SMN2 et qui est administré par voie intrathécale (ponction lombaire). Le mode d’administration n’est pas la seule raison de l’encadrement de traitement. Sa prescription et son administration sont du ressort des centres de référence neuromusculaires [4,5].
Zolgensma® (onasemnogene abeparvovec)
Le Zolgensma® est un médicament de thérapie génique qui traite le défaut moléculaire à l’origine de la maladie. Il agit en introduisant une copie fonctionnelle du gène SMN1 via une perfusion intraveineuse unique d’une solution contenant un vecteur viral de type AAV (Adeno-Associated Virus) et un transgène. Le traitement doit être administré en milieu hospitalier après accord d’une instance nationale (Réunion de Concertation Pluridisciplinaire ou RCP). Douze centres en France ont un agrément des autorités sanitaires pour pratiquer ces injections [6].
Evrysdi® (ridsiplam)
L’Evrysdi® a obtenu une AMM en Europe en mars 2021. Il s’agit d’une petite molécule qui module la maturation de l’ARN messager de SMN2 pour réintégrer l’exon 7 manquant, permettant ainsi la synthèse d’une protéine SMN entière et fonctionnelle. Le traitement est administré par voie orale ou par sonde d’alimentation une fois par jour, à domicile.
La mise en place d’un registre des patients atteints de SMA a été demandée par la Commission de Transparence de la Haute Autorité de Santé (HAS) en date du 31 janvier 2018 pour disposer de données en vie réelle, en complément de celles des essais cliniques réalisés pour ces traitements [4]. Ce registre doit inclure l’ensemble des patients traités et non traités, permettant un recueil d’informations sur les caractéristiques de la maladie par type, l’évaluation de la fonction motrice et respiratoire, la qualité de vie, la tolérance, la mortalité ainsi que la stratégie thérapeutique employée [7].
Registre SMA France
Mise en place du registre
Le registre SMA France a été mis en place en janvier 2020, d’abord sous la forme d’une étude rétrospective puis en tant qu’entrepôt de données de santé récoltant les données rétrospectives et prospectives des patients.
Le coordonnateur de ce registre national est Susana Quijano-Roy, neuro-pédiatre exerçant au centre de Référence des maladies neuromusculaires de l’hôpital Raymond Poincaré à Garches. L’Unité de Recherche Clinique (URC) APHP – Université Paris-Saclay Ouest (Direction de la Recherche Clinique et de l’Innovation) représente l’Assistance-Publique Hôpitaux de Paris (APHP). Cette institution est le promoteur et le responsable du registre et du traitement des données qui y sont entreposées et réutilisées dans le cadre de projets de recherche scientifique et médicale.
Le registre fait l’objet d’un partenariat financier avec l’industrie pharmaceutique dans un cadre légal satisfaisant aux devoirs de protection des données individuelles. Ces financements font l’objet d’une charte de bonnes pratiques et d’une contractualisation, le produit d’exploitation du registre étant à 70 % reversé aux centres participants, le plus souvent sous la forme de personnels dédiés (attaché de recherche clinique [ARC], ou technicien d’études cliniques [TEC]).
Objectifs
La vocation de ce registre est d’obtenir une meilleure connaissance de la pathologie (épidémiologie, histoire naturelle, impact de la maladie, effets des interventions et des traitements) et de répondre au besoin de données en vie réelle compte tenu de l’arrivée de thérapies innovantes ciblant la SMA. Un des objectifs est d’essayer d’identifier les meilleures stratégies thérapeutiques et d’améliorer la prise en charge de ces patients.
Méthode
Le registre inclus tous les patients atteints de SMA liée au gène SMN1 génétiquement confirmés de type 1, 2, 3 et 4, traités ou non, ayant un suivi régulier dans un des centres spécialisés du réseau neuromusculaire national (FILNEMUS) avec au moins une visite à partir du 1er septembre 2016. Les patients ou leurs représentants doivent signer un formulaire de consentement pour pouvoir être inclus dans le registre. Les patients ayant un autre type d’amyotrophie spinale ne sont pas concernés par le registre.
La population cible est estimée à 1 000 patients, dont 50 % de patients pédiatriques, le suivi et la collecte des données s’étendant de janvier 2020 à janvier 2030. Ce recueil de données concerne trois sous-cohortes de populations en fonction de la date de diagnostic et du suivi des patients par le centre (une avec des données historiques-rétrospectives des patients inclus en 2016-2019 et n’ayant plus de suivi ; l’autre historique-prospective pour les patients diagnostiqués avant la participation du centre et ayant encore un suivi et la troisième, prospective pour les patients diagnostiqués après la date de participation du centre). Aucun acte n’est comptabilisé en plus lors de cette participation au registre [8]. À la demande de la HAS, le registre inclura des échelles et données de qualité de vie et d’autonomie, et des données sur le ressenti des patients et de leurs aidants concernant l’efficacité des traitements.
Sur l’ensemble du territoire français, 58 centres ont donné leur accord pour participer au registre, parmi lesquels 31 services pédiatriques et 29 services d’adultes. À fin septembre 2021, 51 centres étaient ouverts, dont 29 services pédiatriques et 24 services d’adultes.
Résultats préliminaires
Au 1er octobre 2021, 666 patients ont été inclus dans la base dont 357 (54 %) enfants et 309 (46 %) adultes dans les 44 des 51 centres participants ouverts. Parmi ces patients, 59 sont décédés.
L’âge des patients varie de 6 mois à 76 ans. La répartition des classes d’âge est définie dans la Figure 1.
 |
Figure 1. Répartition de l’âge des patients inclus dans le registre SMA France au 01/10/2021. |
La répartition du type de SMA est représentative des données de la littérature, avec 22 % de patients de type 1, 42 % de patients de type 2, 35 % de patients de type 3 et 1 % de patients de type 4, comme détaillé dans la Figure 2.
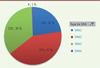 |
Figure 2. Répartition par type de patients inclus dans le registre SMA France au 01/10/2021. |
Le nombre de copie SMN2 dans la population totale des SMA et chez les patients de type 1 est décrite dans les Figures 3 et 4.
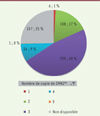 |
Figure 3. Répartition du nombre de copie de SMN2 parmi les patients inclus dans le registre SMA France au 01/10/2021. |
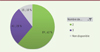 |
Figure 4. Répartition du nombre de copie de SMN2 parmi les patients de type 1 inclus dans le registre SMA France au 01/10/2021. |
Le registre souhaite suivre le nombre de patients traités par une thérapie spécifique de la SMA, ainsi que l’ensemble des patients non traités.
Jusqu’au 1er octobre 2021, 373 patients dans le registre ont reçu au moins un traitement spécifique de la SMA et 231 patients n’ont jamais reçu de traitement spécifique.
Discussion
Un an et demi après son démarrage, le registre SMA a atteint plus de la moitié de l’objectif cible de la population à inclure, malgré les difficultés liées à la crise sanitaire. La qualité et la quantité des données saisies par patient connaissent une amélioration constante grâce à l’implication de tous les acteurs. L’implication des médecins prenant en charge les patients est indispensable pour optimiser la qualité des données colligées par les TEC et les ARC et leur exploitation statistique.
Les données renseignées dans le registre sont analysées lors du rapport annuel partagé avec un comité de pilotage (COPIL) et les partenaires industriels qui sont engagés dans la recherche ou le développement de thérapeutiques et qui soutiennent financièrement le registre.
Le registre SMA a également pour vocation de permettre aux médecins et chercheurs qui le souhaitent, de réaliser des analyses spécifiques à partir des données présentes dans le registre. Il facilitera également la mise en place et le déroulement d’essais cliniques en fournissant des hypothèses de recherche, des données sur les capacités de recrutement des centres, sur la faisabilité, sur les caractéristiques des patients et les pratiques locales des centres.
Conclusion
La collecte nationale des données de l’ensemble de la population SMA et les analyses qui en découleront vont permettre d’obtenir une meilleure connaissance de cette pathologie, ainsi que d’identifier les meilleures stratégies thérapeutiques et d’améliorer conséquemment la prise en charge des malades.
Renseignements pratiques disponibles sur le site du Registre SMA France. http://www.urcpo.pifo.uvsq.fr/SMA/
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Références
- Les Cahiers d’Orphanet. Prévalence des maladies rares : données bibliographiques. Ser Mal Rares [Internet]. janv 2021;(2). www.orpha.net [Google Scholar]
- Iftikhar M, Frey J, Shohan MJ, Malek S, Mousa SA. Current and emerging therapies for Duchenne muscular dystrophy and spinal muscular atrophy. Pharmacol Ther 2021; 220 : 107719. [CrossRef] [PubMed] [Google Scholar]
- Sansone VA, Racca F, Ottonello G, Vianello A, Berardinelli A, Crescimanno G, et al. 1st Italian SMA family association consensus meeting: management and recommendations for respiratory involvement in spinal muscular atrophy (SMA) types I-III, Rome, Italy, 30–31 January 2015. Neuromuscul Disord 2015 ; 25 : 979–989. [CrossRef] [PubMed] [Google Scholar]
- Haute Autorité de Santé. Avis de la Commission de la Transparence - SPINRAZA [Internet]. 2018 janv. (www.has-sante.fr). [Google Scholar]
- Haute Autorité de Santé. Avis de la Commission de la Transparence - SPINRAZA [Internet]. 2020 juill. (https://www.has-sante.fr). [Google Scholar]
- Haute Autorité de Santé. Avis de la Commission de la Transparence - ZOLGENSMA [Internet]. 2020 déc. www.has-sante.fr. [Google Scholar]
- Haute Autorité de Santé. Avis de la Commission de la Transparence - EVRYSDI [Internet]. 2021 sept. (www.has-sante.fr). [Google Scholar]
- Haute Autorité de Santé. Amyotrophie spinale infantile [Internet]. Haute Autorité de Santé. [cité 4 Oct 2021] (www.has-sante.fr). [Google Scholar]
Liste des tableaux
Liste des figures
|
Figure 1. Répartition de l’âge des patients inclus dans le registre SMA France au 01/10/2021. |
| Dans le texte | |
|
Figure 2. Répartition par type de patients inclus dans le registre SMA France au 01/10/2021. |
| Dans le texte | |
|
Figure 3. Répartition du nombre de copie de SMN2 parmi les patients inclus dans le registre SMA France au 01/10/2021. |
| Dans le texte | |
|
Figure 4. Répartition du nombre de copie de SMN2 parmi les patients de type 1 inclus dans le registre SMA France au 01/10/2021. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.