")
")
| Issue |
Med Sci (Paris)
Volume 21, Décembre 2005
Métabolisme glucidolipidique et risque cardiovasculaire: Nouvelle approches
|
|
|---|---|---|
| Page(s) | 16 - 19 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/20052111s16 | |
| Published online | 15 novembre 2005 | |
Les maladies lysosomales : mécanismes pathologiques et options thérapeutiques
Lysosomal storage diseases: pathological mechanisms and therapeutic options
Department of Biological Chemistry, Weizmann Institute of Science, Rehovot 76100, Israël
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Plus de 40 maladies lysosomales sont actuellementconnues et répertoriées en catégories selonla nature des métabolites accumulés. Dans cetarticle sont évoqués les troubles enzymatiqueset la symptomatologie des principales maladiesest décrite. Les possibilités de traitementsont discutées, en particulier le traitement parenzyme de substitution qui est actuellement leplus efficace dans certaines de ces maladies desurcharge.
Abstract
More than 40 lysosomal disorders are known and classifiedinto broad categories based on the nature of the undegradedmetabolites. In this review, the defective enzymatic activitiesare analyzed, and the pathology of the disorders are described.Various treatments are discussed, including enzyme replacementtherapy, which is the most successful treatment availabletoday.
© 2005 médecine/sciences - Inserm / SRMS
Les maladies de surcharge lysosomales (lysosomal storage disorders, LSD) sont des affections monogéniques dues à des troubles de l’activité des protéines des lysosomes, entraînant une accumulation intralysosomale de métabolites non dégradés, communément appelés produits de surcharge. On en connaît plus d’une quarantaine qui se répartissent en grandes catégories : sphingolipidoses, oligosaccharidoses et mucopolysaccharidoses, selon la nature du matériel accumulé [1]. Les sphingolipidoses, ou maladies de surcharge des sphingolipides (SL) sont dues, dans la plupart des cas, à une diminution de l’activité catalytique d’une ou de plusieurs hydrolases lysosomales intervenant à l’une des étapes de la dégradation des SL, et conduisant à une accumulation intracellulaire de SL non degradés [2]. Par exemple, la maladie de Gaucher, une des LSD les plus fréquentes, est causée par des mutations du gène codant la β-glucosidase acide (ou glucocérébrosidase), ce qui entraîne une accumulation de glucosylcéramide (GlcCer) intralysosomale. La maladie de Fabry est la conséquence d’une activité défectueuse de l’α-galactosidase A, résultant en une accumulation de globotriaosylcéramide (Gb3). Les voies métaboliques dont la perturbation aboutissent aux différentes sphingolipidoses sont représentées dans la Figure 1.
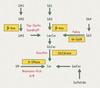 |
Figure 1. Voies de dégradation des glycosphingolipides (GSL). Les enzymes défectueuses dans ces maladies sont encadrées en jaune et le nom de la maladie est en rouge. β-Hex: β-hexosaminidase; α-Gal A: α-galactosidase A ; GlcCérase: glucocérébrosidase encore appelée β-glucosidase acide ; A-Smase: sphingomyélinase acide. |
Bien que les bases génétiques et moléculaires des maladies lysosomales aient été étudiées depuis des années, on connaît mal les mécanismes qui conduisent de la surcharge lysosomale aux manifestations cliniques. Certes, depuis la découverte des lysosomes, dénommés suicide bags par C. De Duve et ses collaborateurs, un grand intérêt leur a été porté, en particulier dans les maladies qui résultent d’une incapacité des enzymes lysosomales à fonctionner efficacement [3]. La littérature scientifique est pourtant peu diserte sur les voies biochimiques en aval, ainsi que sur les voies secondaires, responsables d’un dysfonctionnement cellulaire et tissulaire, et par conséquent sur les véritables mécanismes pathologiques. En d’autres termes, pour quelles raisons l’accumulation intracellulaire ou intralysosomale de l’un ou l’autre de ces produits de surcharge entraîne-t-elle un dysfonctionnement, sans qu’il y ait corrélation apparente entre le type de substrat qui s’accumule et l’évolution de la maladie ? Les manifestations cliniques des LSD sont très hétérogènes, progressives, et le rythme de l’évolution varie beaucoup d’une maladie à l’autre et même au sein d’une même maladie. On ignore les raisons de cette hétérogénéité mais il est probable qu’elle dépend à la fois de la quantité et de la nature du matériel de surcharge et des voies biochimiques et cellulaires qui sont altérées (Figure 2). Cependant, très peu de liens directs ont été mis en évidence jusqu’à présent entre l’évolution clinique, la pathologie, et la biochimie des ces maladies.
 |
Figure. 2. Pathogénie des LSD. Les LSD se caractérisent toutes par une accumulation de matériel de surcharge intralysosomal and intracellulaire qui est la cause primaire de la maladie. Mais le large éventail des symptômes montre que de nombreuses voies secondaires biochimiques et cellulaires doivent être activées. Ainsi, les métabolites qui s’accumulent ont des conséquences biochimiques et affectent des voies cellulaires, provoquant des lésions tissulaires, une altération de l’expression des gènes et une activation de voies tertiaires qui restent encore mal connues. |
Pathologie des LSD
À l’exception de la maladie de Gaucher de type 1, dont la symptomatologie est exclusivement viscérale, les deux tiers des LSD comportent une atteinte neurologique et, dans le cas des SL, cette atteinte est manifeste. L’implication neurologique au cours des glycosphingolipidoses (GSL) n’est pas surprenante car les GSL jouent un rôle essentiel dans le système nerveux central où ils sont présents en grande quantité. Pourtant, jusqu’à une date récente, les connexions entre l’accumulation des GSL et la progression de la maladie étaient encore peu connues. Depuis 4 ou 5 ans toutefois, de réels progrès ont été faits, grâce à la découverte, dans au moins quatre maladies de surcharge de SL, de l’atteinte de la voie de l’homéostasie du Ca2+ intracellulaire [4].
Les premiers travaux montrèrent que les neurones qui accumulaient le glycosylcéramide (GlcCer), observés dans les formes de maladie de Gaucher avec atteinte neurologique et traitées par la caféine, libéraient plus de Ca2+ à partir des compartiments intracellulaires que les cellules non traitées. Cette libération du Ca2+ résultait d’une augmentation de la sensibilité aux agents neurotoxiques [5, 6]. Puisque la caféine est un agoniste du récepteur de la ryanodine (RyaR), un canal calcique localisé dans le réticulum endoplasmique (RE), il a été montré que l’accumulation de GlcCer dans les neurones augmentait la libération du Ca2+ induit par les agonistes à partir du RE via le RyaR [7], avec mort cellulaire ultérieure. De plus, l’étude de la libération du Ca2+ in vitro à partir des microsomes démontrait que seul, parmi tous les sphingolipides testés, le GlcCer était capable de stimuler la libération du Ca2+ induite par les agonistes [7]. Une étude sur des coupes de cerveaux humains de patients atteints de maladie de Gaucher, obtenus post-mortem montrait une corrélation entre les taux de GlcCer, ceux de Ca2+ et le phénotype de la maladie [8]. Ces constatations suggéraient qu’un trouble de l’homéostasie du Ca2+ pouvait être un des mécanismes à l’origine de la physiopathologie des formes neuropathiques de maladie de Gaucher.
Nous avons aussi observé une perturbation de l’homéostasie du Ca2+ dans un modèle murin de maladie de Sandhoff, la souris Hexb−/−, bien que le mécanisme soit différent de celui observé dans la maladie de Gaucher. Dans les microsomes obtenus à partir de souris Hexb−/−, le taux de prise du Ca2+, via la sarco/endoplasmic reticulum Ca2+ ATPase (SERCA), était considérablement réduit, bien qu’aucune différence dans le taux de libération du Ca2+ via le RyaR n’ait été observée [9]. L’activité réduite de la SERCA (sarco/endoplasmic reticulum Ca2+ ATPase) était inversée quand les souris Hexb−/− étaient nourries avec de la N-butyldésoxynojirimycine, inhibiteur de la synthèse des glycolipides (voir plus loin), qui réduit les taux de conservation de GM2 [10], en corrélation avec : (1) une réduction des quantités de GM2 accumulées ; (2) un retard d’apparition des symptômes ; et (3) une augmentation de la durée de vie de ces souris [11].
Nous avons aussi récemment montré que l’homéostasie du Ca2+ est perturbée dans un modèle murin de maladie de Niemann-Pick A [12], la souris asm, invalidée pour le gène de la sphingomyélinase acide. Cependant, le mécanisme responsable du trouble de l’homéostasie du Ca2+ est complètement différent de celui des autres modèles : les taux d’expression de la protéine SERCA étaient significativement réduits dans le cerveau des souris asm−/−, de même que les taux du récepteur de l’inositol 1, 4, 5-triphosphate, principal canal calcique dans le cervelet. Ces résultats suggèrent qu’une perturbation de l’homéostasie du Ca2+ joue un rôle au cours de la neurodégénérescence observée dans une population spécifique de cellules de Purkinje dans la maladie de Niemann-Pick A, sans que soient identifiés les lipides qui entraînent la mort cellulaire. Enfin, une étude récente, dans un modèle murin de gangliosidose à GM1 [13], a démontré que la mort neuronale provoquée par l’accumulation de GM1 dans le RE résultait d’une déplétion en Ca2+ et d’une activation consécutive de la réponse des protéines mal repliées.
Ainsi, ces dernières années, nous avons assisté à un début de caractérisation des voies impliquées dans les processus pathologiques observés dans les LSD. Toutefois, il est probable qu’il existe d’autres voies additionnelles, et le défi dans ce domaine est de parvenir à une analyse détaillée des événements conduisant de l’accumulation dans les lysosomes d’un matériel de surcharge à un dysfonctionnement puis à la mort cellulaire.
Traitement des LSD
Après avoir constaté que les LSD étaient dues à un défaut d’activité des enzymes lysosomales, C. de Duve a exprimé sa profonde satisfaction de voir que ces découvertes, non seulement enrichissaient les connaissances, mais pourraient contribuer à vaincre un jour ces maladies [3]. Dans la troisième partie de cet article, les possibilités présentes et futures de traitement des LSD seront discutées.
Théoriquement, les options thérapeutiques pour les LSD devraient viser à remplacer soit le gène, soit l’enzyme déficiente, ou encore cibler les systèmes atteints par une accumulation de produits de surcharge [1]. Le traitement idéal pour les LSD, et pour toutes les maladies métaboliques héréditaires serait évidemment une thérapie génique somatique. Mais la probabilité d’une telle thérapie dans un futur proche reste incertaine.
Le traitement le plus efficace aujourd’hui est la thérapie substitutive par remplacement enzymatique (enzyme replacement therapy, ERT) avec toutefois un problème important : comment adresser l’enzyme aux cellules déficientes ? Dans la maladie de Gaucher, pour laquelle un traitement fut possible dès 1991, la délivrance aux macrophages de la β-glucosidase acide initialement extraite du placenta (alglucérase) fut obtenue en remodelant les chaînes latérales d’oligosaccharides pour exposer leurs résidus mannose. Après liaison aux récepteurs mannose présents à la surface des macrophages, l’enzyme, aujourd’hui produite par technologie recombinante, est délivrée aux lysosomes par endocytose [14]. Sur le même modèle, une autorisation de mise sur le marché a été accordée pour la maladie de Fabry et très récemment pour les mucopolysaccharidoses (MPS) de type I (maladie de Hurler-Scheie) et VI (maladie de Maroteaux-Lamy). Pour d’autres maladies lysosomales (MPS II, Niemann-Pick type B et glycogénose de type II) des essais précliniques ou cliniques sont en cours [15].
Des essais ont été réalisés pour améliorer l’efficacité des ERT [16]. Ceux-ci comportent un meilleur ciblage des cellules atteintes, et une amélioration de la stabilité ou de l’efficacité des enzymes recombinantes. Pour ce faire, la connaissance de la structure tridimensionnelle de l’enzyme concernée est essentielle. Dans la maladie de Gaucher, la structure de la glucocérébrosidase acide vient précisément d’être déterminée [17, 18]. La production d’une glucocérébrosidase plus stable ou plus active pourrait réduire le nombre de perfusions et ainsi diminuer le coût du traitement. La connaissance de la structure de la glucocérébrosidase a par ailleurs permis une approche plus rationnelle de la production de petites molécules pouvant être utilisées comme chaperons thérapeutiques [19].
D’autres traitements pour les LSD sont fondés sur la prévention des anomalies métaboliques et cellulaires qui surviennent lors de l’accumulation de produits de surcharge, ou sur l’inhibition partielle de leur synthèse, freinant ainsi leur accumulation. Cette dernière stratégie a été essayée pour les sphingolipidoses, dans lesquelles l’inhibition de la synthèse de GSL par la N-butyldéosynojirimycine (inhibiteur de la glycosylcéramide synthase, première enzyme de la voie de synthèse des GSL) améliore les symptômes chez les patients atteints de maladie de Gaucher [20]. Ce type de traitement, connu comme substrate deprivation ou inhibition de synthèse pourrait légitimement être essayé dans d’autres sphingolipidoses où les GSL s’accumulent dans le système nerveux central puisque les petites molécules inhibitrices sont capables de traverser la barrière hémato-encéphalique à la différence des enzymes recombinantes. Cependant, le manque d’études au long cours sur l’effet de la déplétion des GSL, en particulier au cours du développement, limite encore fortement cette approche.
Dans tous les cas, une meilleure connaissance des mécanismes moléculaires et cellulaires présidant, au-delà de la simple surcharge métabolique, à la physiopathologie des maladies lysosomales, sera une étape clé pour le développement de nouvelles options thérapeutiques.
Références
- Futerman AH, Van Meer G. The cell biology of lysosomal storage disease. Nat Rev Mol Cell Biol 2004; 5 : 554–65. [Google Scholar]
- Raas-Rothschild A, Pankova-Kholmyansky I, Kacher Y, Futerman AH. Glycosphingolipidoses: beyond the enzymatic defect. Glycoconj J 2004 ; 21 : 295–304. [Google Scholar]
- de Duve C. Exploring cells with a centrifuge. Science 1975; 189 : 186–94. [Google Scholar]
- Ginzburg L, Kacher Y, Futerman AH. The pathogenesis of glycosphingolipid storage disorders. Semin Cell Dev Biol 2004; 5 : 417–31. [Google Scholar]
- Korkotian E, Schwarz A, Pelled D, et al. Elevation of intracellular glucosylceramide levels results in an increase in endoplasmic reticulum density and in functional calcium stores in cultured neurons. J Biol Chem 1999; 274 : 21673–8. [Google Scholar]
- Pelled D, Shogomori H, Futerman AH. The increased sensitivity of neurons with elevated glucocerebroside to neurotoxic agents can be reversed by imiglucerase. J Inherit Metab Dis 2000; 23 : 175–84. [Google Scholar]
- Lloyd-Evans E, Pelled D, Riebeling C, et al. Glucosylceramide and glucosylsphingosine modulate calcium mobilization from brain microsomes via different mechanisms. J Biol Chem 2003 ; 278 : 23594–9. [Google Scholar]
- Pelled D, Trajkovic-Bodennec S, Lloyd-Evans E, Sidransky, E, Schiffmann R, Futerman AH. Enhanced calcium release in the acute neuronopathic form of Gaucher disease. Neurobiol Dis 2005; 18 : 83–8. [Google Scholar]
- Pelled D, Lloyd-Evans E, Riebeling C, et al. Inhibition of calcium uptake via the sarco/endoplasmic reticulum Ca2+-ATPase in a mouse model of Sandhoff disease and prevention by treatment with N-butyldeoxynojirimycin. J Biol Chem 2003; 278 : 29496–501. [Google Scholar]
- Lachmann RH. Miglustat. Oxford GlycoSciences/Actelion. Curr Opin Investig Drugs 2003; 4 : 472–9. [Google Scholar]
- Jeyakumar M, Butters TD, Cortina-Borja M, et al. Delayed symptom onset and increased life expectancy in Sandhoff disease mice treated with N-butyldeoxynojirimycin. Proc Natl Acad Sci USA 1999; 96 : 6388–93. [Google Scholar]
- Ginzburg L, Futerman AH. Defective calcium homeostasis in the cerebellum in a mouse model of Niemann-Pick A disease. J Neurochem 2005 (sous presse). [Google Scholar]
- Tessitore A, del P Martin M, Sano R, et al. GM1-ganglioside-mediated activation of the unfolded protein response causes neuronal death in a neurodegenerative gangliosidosis. Mol Cell 2004; 15 : 753–66. [Google Scholar]
- Grabowski GA, Hopkin RJ. Enzyme therapy for lysosomal storage disease: principles, practice, and prospects. Ann Rev Genom Hum Genet 2003; 4 : 403–36. [Google Scholar]
- Desnick RJ, Schuchman EH. Enzyme replacement and enhancement therapies: lessons from lysosomal disorders. Nat Rev Genet 2002; 3 : 954–66. [Google Scholar]
- Futerman AH, Sussman JL, Horowitz M, et al. New directions in the treatment of Gaucher disease. Trends Pharmacol Sci 2004; 25 : 147–51. [Google Scholar]
- Dvir H, Harel M, McCarthy AA, et al. X-ray structure of human acid-beta-glucosidase, the defective enzyme in Gaucher disease. EMBO Rep 2003; 4 : 704–9. [Google Scholar]
- Premkumar L, Sawkar AR, Boldin-Adamsky S, et al. X-ray structure of human acid-beta-glucosidase covalently bound to conduritol-B-epoxide. Implications for Gaucher disease. J Biol Chem 2005; 280 : 23815–9. [Google Scholar]
- Fan JQ. A contradictory treatment for lysosomal storage disorders: inhibitors enhance mutant enzyme activity. Trends Pharmacol Sci 2003; 24 : 355–60. [Google Scholar]
- Cox T, Lachmann R, Hollak C, et al. Novel oral treatment of Gaucher’s disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet 2000; 355 : 1481–5. [Google Scholar]
Liste des figures
|
Figure 1. Voies de dégradation des glycosphingolipides (GSL). Les enzymes défectueuses dans ces maladies sont encadrées en jaune et le nom de la maladie est en rouge. β-Hex: β-hexosaminidase; α-Gal A: α-galactosidase A ; GlcCérase: glucocérébrosidase encore appelée β-glucosidase acide ; A-Smase: sphingomyélinase acide. |
| Dans le texte | |
|
Figure. 2. Pathogénie des LSD. Les LSD se caractérisent toutes par une accumulation de matériel de surcharge intralysosomal and intracellulaire qui est la cause primaire de la maladie. Mais le large éventail des symptômes montre que de nombreuses voies secondaires biochimiques et cellulaires doivent être activées. Ainsi, les métabolites qui s’accumulent ont des conséquences biochimiques et affectent des voies cellulaires, provoquant des lésions tissulaires, une altération de l’expression des gènes et une activation de voies tertiaires qui restent encore mal connues. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.