")
")
Figure 1.
Télécharger l'image originale
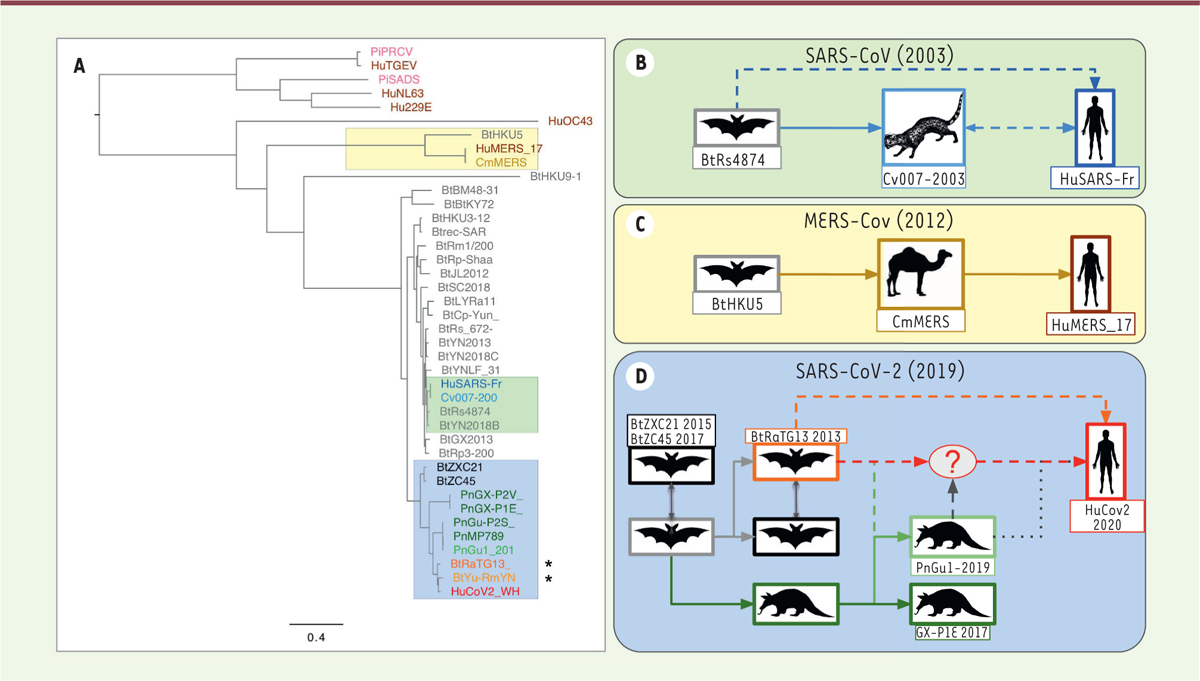
Phylogénie et émergence des coronavirus. A. Arbre de génomes complets de coronavirus, en se fondant sur un alignement multiple (clustalw) suivi d’une inférence en maximum de vraisemblance (PhyML). Les génomes assemblés à partir de données métagénomiques sont marqués d’une étoile. Le préfixe des virus correspond aux espèces: Bt (chauve-souris), Hu (humain), Pn (pangolin), Cv (civette), Cm (dromadaire), Pi (porc). On constate que les distances entre HuCoV2 et les souches virales les plus proches (BtYuRmYN02[r>t], BtRaTG13) sont plus élevées que pour SARS-CoV (humain - civette) ou MERS-CoV (humain-dromadaire) (B-D) Hypothèses de transmission du réservoir animal (chauve-souris) jusqu’à l’homme, fondées sur la phylogénie moléculaire des génomes viraux. B.Pour la pandémie SARS-Cov de 2003, l’hôte intermédiaire est la civette. Une transmission directe de la chauve-souris à l’homme est également envisagée. C. Pandémie MERS-CoV de 2012, avec le dromadaire comme hôte intermédiaire. Plusieurs événements de transmission directe ont été documentés. D. Pandémie Covid-19. Plusieurs scénarios sont proposés concernant le dernier hôte avant la transmission à l’homme.
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.