")
")
Figure 2.
Télécharger l'image originale
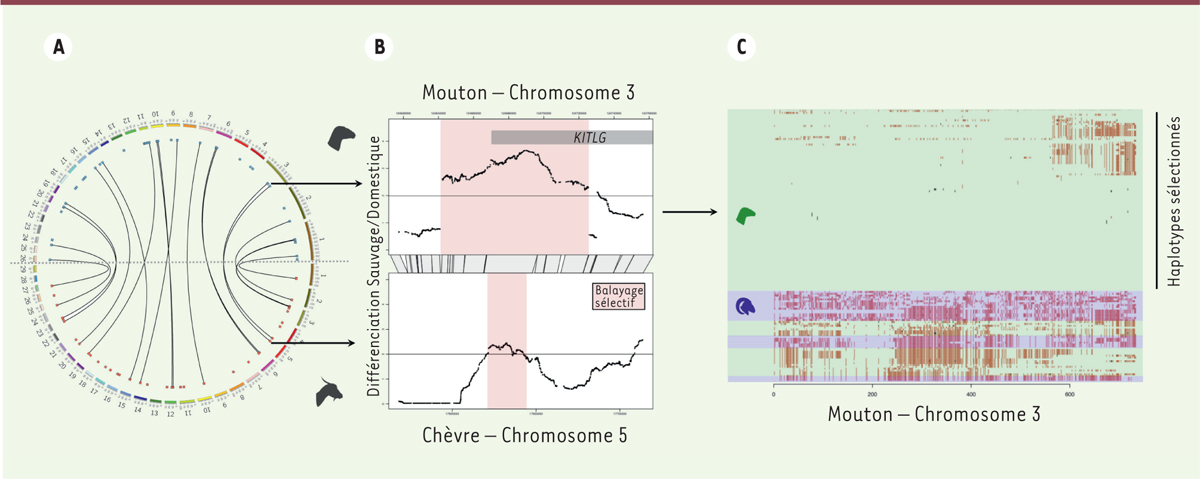
Caractérisation des régions génomiques influencées par la domestication chez la chèvre et le mouton. A. Localisation des signatures de sélection liées à la domestication. Chaque tiret de couleur numéroté représente un chromosome de mouton (moitié supérieure) ou de chèvre (moitié inférieure) ; les points rouge et bleu localisent les régions sélectionnées qui sont reliées lorsqu’elles sont homologues entre les deux espèces. B. Exemple de deux régions homologues présentant un balayage sélectif. La différenciation entre groupes domestique et sauvage le long de la portion de chromosome est estimée en fonction du taux de détection de faux positifs (false discovery rate) exprimé en -Log10 FDR-val-q (la valeur -q est l’équivalent de la valeur p des tests statistiques dans une approche de faux positifs), considéré significatif au seuil de val-q = 10-2, soit un pourcentage de faux positifs de 1 %. Le balayage sélectif inclue une portion du gène KITLG (KIT proto-oncogene receptor tyrosine kinase ligand) dont la position est représentée par une barre grisée. C..Haplotypes de moutons dans la portion sous sélection. Chaque ligne représente un individu (bleu : sauvage, vert : domestique), l’alternance des couleurs représente la variation de la séquence d’ADN par rapport au génome de référence du mouton et les points noirs représentent les données manquantes. Les haplotypes sont ordonnés selon la proximité de leurs séquences.
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.