")
")
| Numéro |
Med Sci (Paris)
Volume 29, Numéro 4, Avril 2013
|
|
|---|---|---|
| Page(s) | 361 - 364 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/2013294009 | |
| Publié en ligne | 26 avril 2013 | |
Aldéhydes et anémie de Fanconi
L’ennemi de l’intérieur
Aldehydes and Fanconi anaemia: the enemy within
Medical Research Council, Laboratory of Molecular Biology, Francis Crick avenue, Cambridge Biomedical Campus, Cambridge, CB2 0QH, Royaume-Uni
La survie d’un organisme multicellulaire est étroitement liée au maintien de l’homéostasie dans l’ensemble des tissus constituant cet organisme. Cette capacité à maintenir cet équilibre vital est assurée par des populations de cellules souches, via trois caractéristiques principales : (1) la possibilité de se diviser et de renouveler ce compartiment en générant de nouvelles cellules souches ; (2) l’absence de fonctions tissulaires spécifiques ; et (3) la capacité de se différencier en cellules spécialisées. La moelle osseuse, où résident les cellules souches hématopoïétiques (CSH) responsables de la production des cellules du sang, constitue un tissu modèle pour l’étude des cellules souches somatiques. En effet, l’identification de multiples marqueurs de surface et le développement d’anticorps dirigés contre ces marqueurs et utilisables en cytométrie de flux (FACS, fluorescent activated cell sorter), permettent de discriminer et d’isoler les divers précurseurs des cellules sanguines.
Les cellules souches sont sensibles, comme toute cellule, aux conditions de stress, en particulier aux dommages de l’ADN. Ainsi, au cours du temps, l’accumulation de mutations contribue au déclin des capacités des cellules souches. L’existence de mécanismes de réparation de l’ADN est donc essentielle au maintien des populations de cellules souches [1]. Une étude récente de notre groupe décrit le rôle d’un système de réparation de l’ADN et la nature génotoxique de métabolites endogènes dans les CSH de souris [2].
L’anémie de Fanconi, une pathologie s’accompagnant d’un défaut de réparation de l’ADN
La thématique de recherche principale de notre équipe concerne l’étude des mécanismes de réparation de l’ADN et, en particulier, celle des gènes impliqués dans l’anémie de Fanconi. Notre approche est essentiellement génétique et se base sur des modèles cellulaires et murins (souris génétiquement modifiées ou knockout [KO]). Du nom du pédiatre suisse Guido Fanconi qui décrivit cette maladie génétique rare au début du XXe siècle [3], l’anémie de Fanconi se manifeste dès l’enfance par des malformations squelettiques, une forte prédisposition à certains types de cancers (en particulier des leucémies myéloïdes) et une aplasie médullaire progressive. Les cellules de ces patients sont très sensibles à des drogues anticancéreuses, telles que la cisplatine ou la mitomycine C. Cette sensibilité constitue la base du test de diagnostic dit de « cassures chromosomiques » [4]. En effet, en liant de façon covalente deux bases opposées (interbrins d’ADN) ou adjacentes (intrabrin d’ADN), ces drogues induisent des lésions (ponts inter- ou intrabrins, DNA crosslinks) de l’ADN qui bloquent la réplication et la transcription. Ces ponts interbrins ont pour conséquence une augmentation des cassures chromosomiques. Dans les cellules de patients atteints d’anémie de Fanconi, ceci est dû à un défaut de réparation de l’ADN. À ce jour, une quinzaine de gènes ont été identifiés, codant pour des protéines impliquées dans cette voie de réparation des lésions pontantes de l’ADN.
Les aldéhydes, source de dommages à l’ADN
Les cassures chromosomiques se manifestent spontanément dans les cellules de patients, en dehors de la présence de drogues comme la cisplatine, ce qui suggère l’existence d’agents génotoxiques présents de façon naturelle dans les cellules de ces patients. Notre groupe a confirmé cette hypothèse en montrant en 2011 que les aldéhydes produits par le métabolisme cellulaire (en particulier l’acétaldéhyde) étaient à l’origine de dommages à l’ADN dans les cellules de patients atteints d’anémie de Fanconi [5]. Des souris double KO pour les gènes Fancd2 (un des 15 gènes impliqués dans l’anémie de Fanconi [6]) et Aldh2 (le gène codant pour une enzyme qui oxyde l’acétaldéhyde en acétate) développent une leucémie dès l’âge de trois à six mois. Ces animaux naissent avec des malformations squelettiques et oculaires, indiquant que la production spontanée d’acétaldéhyde et son accumulation sont suffisantes pour aggraver le phénotype des souris Fancd2−/−. De plus, l’administration d’éthanol (un précurseur exogène de l’acétaldéhyde) au lieu d’eau à des souris Fancd2−/−Aldh2−/− accélère le développement d’une aplasie qui s’installe en seulement 10 jours.
Plus récemment, nous avons précisé le rôle myélotoxique des aldéhydes dans la progression de cette anémie aplasique [2]. Tout d’abord, nous avons remarqué que certaines souris Fancd2−/−Aldh2−/− survivent sans développer de leucémie, mais succombent à une anémie. Une analyse post mortem a révélé chez ces animaux une accumulation de la forme variante de l’histone 2A (γH2AX, un marqueur de lésions de l’ADN) et de la caspase 3 (un marqueur d’apoptose) dans les cellules de la moelle osseuse. Ces marqueurs attestent de l’accumulation de dommages à l’ADN qui expliquent l’hypocellularité de la moelle osseuse.
Nous avons ensuite déterminé in vitro la toxicité de l’acétaldéhyde sur les différentes populations cellulaires hématopoïétiques. La toxicité de l’acétaldéhyde vis-à-vis des cellules sanguines différenciées (lymphocytes B et T, érythrocytes, etc.) n’est pas majorée par l’association des mutations des gènes Fancd2 et Aldh2 par rapport à des cellules mutées uniquement dans le gène Fancd2. En revanche, les cellules souches/progénitrices de la moelle osseuse de souris Fancd2−/−Aldh2−/−, purifiées par tri cellulaire, sont nettement plus sensibles à l’acétaldéhyde que les cellules souches souches/progénitrices contrôles (issues de simple mutants Aldh2−/− ou Fancd2−/−). L’effet génotoxique de l’acétaldéhyde peut être observé dès l’âge de huit semaines, puisque le nombre de CSH chez les jeunes souris adultes Fancd2−/−Aldh2−/− est diminué d’un facteur 250. Les CSH sont identifiées en cytométrie de flux par l’expression de marqueurs de surface spécifiques : protéines SLAM, c-Kit et Sca-1 (SREBP cleavage activity-1) et par l’absence de marqueurs spécifiques de lignages (Lin-). Cette série d’expériences in vitro démontre ainsi la sensibilité spécifique à l’acétaldéhyde des cellules souches - par opposition aux cellules différenciées - chez les animaux Fancd2−/−Aldh2−/−. De même, il est possible que les lésions de l’ADN causées par les aldéhydes soient majoritairement présentes dans les CSH, où elles sont réparées par l’action des protéines Fanconi.
Aldh et protéines Fanconi, deux systèmes de protection et de réparation de l’ADN
Malgré tout, ces preuves histologiques et quantitatives n’expliquent pas le dysfonctionnement de ces cellules aboutissant à l’anémie aplasique. Afin d’évaluer les capacités régénératrices des CSH Fancd2−/−Aldh2−/− , nous avons utilisé une stratégie de transplantation de moelle osseuse, qui représente le meilleur moyen de tester les fonctions de repopulation des CSH greffées chez un hôte irradié. L’efficacité de reconstitution à long terme du système hématopoïétique de l’hôte est évaluée quatre mois après la greffe par la détection des cellules du donneur par FACS et PCR (polymerase chain reaction) quantitative. Nous avons donc utilisé de jeunes souris adultes (huit semaines, non anémiques et non atteintes de leucémie) comme source de moelle osseuse et donc de CSH, greffées dans des hôtes préalablement irradiés. Le résultat de cette expérience est surprenant : les CSH Fancd2−/−Aldh2−/− ont une capacité de reconstitution de la moelle osseuse extrêmement réduite en comparaison de celle de CSH contrôles (réduction d’un facteur 638). En revanche, la capacité de reconstitution des CSH Aldh2−/− et Fancd2−/− est moins limitée (réduction d’un facteur 2,75 et 38, respectivement) (Figure 1) [7].
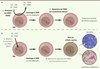 |
Figure 1. Le double système de protection contre l’effet génotoxique des aldéhydes dans le contexte de la réparation de l’ADN. A. L’action enzymatique catabolique des aldéhydes déshydrogénases (Aldh2 ou Aldh) permet de détoxifier la cellule de ces composés très réactifs. Malgré cette première barrière, des quantités résiduelles d’aldéhydes (acétaldéhyde [CH3-CHO] ou autres aldéhydes [R-CHO]) peuvent induire des lésions de l’ADN. Ces lésions sont ensuite réparées par le système des protéines Fanconi. Ce mécanisme de protection à deux niveaux permet ainsi de maintenir l’intégrité du génome des cellules souches hématopoïétiques et la production contrôlée de cellules du sang. B. Dans le cas des souris Fancd2−/−Aldh2−/−, l’accumulation d’aldéhydes non oxydés a pour conséquence une augmentation du nombre de lésions de l’ADN dans les cellules souches hématopoïétiques. Le système de réparation par les protéines Fanconi étant incomplet et défectueux en raison de la mutation du gène Fancd2, ces lésions perdurent et aboutissent à l’émergence de cellules leucémiques ou à l’épuisement du stock de cellules souches hématopoïétiques. Ce modèle explique donc le développement de leucémie et d’anémie aplasique chez les souris Fancd2−/−Aldh2−/−. CSH : cellule souche hématopoïétique ; Aldh2 : aldéhyde déshydrogénase 2 ; Aldh : aldéhydes déshydrogénases ; CH3-CHO : acétaldéhyde ; R-CHO : aldéhydes. |
Ces résultats démontrent que le catabolisme des aldéhydes dans les CSH est un mécanisme important de protection, agissant de concert avec le système de réparation de l’ADN impliquant les protéines Fanconi (Figure 1). L’approche génétique consistant à cibler un gène, tel que l’Aldh2, dans un modèle murin de l’anémie de Fanconi (souris Fancd2−/− ) permet d’identifier une source endogène d’agents génotoxiques et de reproduire plus fidèlement l’évolution de la maladie, telle qu’elle est observée chez les patients. En effet, à ce jour, la plupart des modèles murins de l’anémie de Fanconi, à l’exception récente des souris Slx4−/− (structure-specific endonuclease subunit homolog), ne miment pas l’insuffisance médullaire, pourtant un des symptômes caractéristiques de la maladie humaine [8, 9]. La capacité extrêmement réduite des CSH de jeunes souris Aldh2−/−Fancd2−/− à reconstituer des hôtes irradiés démontrent l’aspect progressif de l’insuffisance médullaire. En effet, à l’âge de huit semaines, ces souris donneuses ne sont ni leucémiques ni anémiques, mais la fonction de leurs CSH est déjà altérée. Ces observations sont en accord avec une récente étude de la pathogénie de l’insuffisance médullaire chez les patients atteints d’anémie de Fanconi [10]. R. Ceccaldi et al. [10] ont montré que l’hypocellularité de la moelle osseuse et le déficit fonctionnel des cellules souches/progénitrices précèdent l’expression clinique de la maladie ; elles seraient en relation avec une activation excessive de l’axe p53/p21 (entraînant un arrêt du cycle cellulaire) en réponse aux dommages de l’ADN non réparés et au stress cellulaire endogène. Ces auteurs ont démontré que des progéniteurs hématopoïétiques de sang de cordon ombilical humain normal, traités par un siARN ciblant FANCD2 sont incapables de reconstituer des souris immunodéficientes Nod/Scid/IL2-Rγc nulles irradiées. L’extinction concomitante de P53 restaure ce potentiel de reconstitution. La précocité de ces anomalies chez les patients atteints d’anémie de Fanconi suggère que le défaut de réparation de l’ADN responsable de l’insuffisance médullaire émerge dès le développement intra-utérin. Il est probable que les aldéhydes soient aussi impliqués dans la génotoxicité au même stade de la vie (Figure 1).
Conclusion
D’un point de vue thérapeutique, un modèle tel que celui des souris Aldh2−/−Fancd2−/− pourrait être utilisé dans des projets visant à optimiser des traitements de l’insuffisance médullaire chez les patients atteints d’anémie de Fanconi. Il est aussi envisageable de tester l’effet génotoxique d’autres métabolites cellulaires, en introduisant des mutations dans les gènes impliqués dans des mécanismes de détoxification, codant pour des enzymes équivalentes aux aldéhydes déshydrogénases (Aldh). D’un point de vue fondamental, il reste à déterminer si l’effet des aldéhydes sur l’ADN est direct ou indirect, la nature exacte des lésions induites par les aldéhydes, et le mécanisme biochimique impliqué dans la réparation des lésions pontantes de l’ADN.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Remerciements
Le travail cité dans cette nouvelle a été financé par le Medical research council (KJP), le Cancer research UK (GPC), le Homerton college Cambridge (GPC), le Milstein fund (JIG), le Darwin trust of Edinburgh (JIG) et la March of dimes foundation (FPML). Merci à Hayat Arzouk et Guillaume Guilbaud pour leurs commentaires sur ce manuscrit.
Références
- Niedernhofer LJ. DNA repair is crucial for maintaining hematopoietic stem cell function. DNA Repair (Amst) 2008 ; 7 : 523–529. [CrossRef] [PubMed] [Google Scholar]
- Garaycoechea JI, Crossan GP, Langevin F, et al. Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature 2012 ; 489 : 571–575. [CrossRef] [PubMed] [Google Scholar]
- Lobitz S, Velleuer E. Guido Fanconi (1892–1979): a jack of all trades. Nat Rev Cancer 2006 ; 6 : 893–898. [CrossRef] [PubMed] [Google Scholar]
- Auerbach AD, Adler B, Chaganti RS. Prenatal and postnatal diagnosis and carrier detection of Fanconi anemia by a cytogenetic method. Pediatrics 1981 ; 67 : 128–135. [PubMed] [Google Scholar]
- Langevin F, Crossan GP, Rosado IV, et al. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 2011 ; 475 : 53–58. [CrossRef] [PubMed] [Google Scholar]
- Houghtaling S, Timmers C, Noll M, et al. Epithelial cancer in Fanconi anemia complementation group D2 (Fancd2) knockout mice. Genes Dev 2003 ; 17 : 2021–2035. [CrossRef] [PubMed] [Google Scholar]
- Parmar K, Kim J, Sykes SM, et al. Hematopoietic stem cell defects in mice with deficiency of Fancd2 or Usp1. Stem Cells 2010 ; 28 : 1186–1195. [CrossRef] [PubMed] [Google Scholar]
- Parmar K, D’Andrea A, Niedernhofer LJ. Mouse models of Fanconi anemia. Mutat Res 2009 ; 668 : 133–140. [CrossRef] [PubMed] [Google Scholar]
- Crossan GP, van der Weyden L, Rosado IV, et al. Disruption of mouse Slx4, a regulator of structure-specific nucleases, phenocopies Fanconi anemia. Nat Genet 2011 ; 43 : 147–152. [Google Scholar]
- Ceccaldi R, Parmar K, Mouly E, et al. Bone marrow failure in Fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell 2012 ; 11 : 36–49. [CrossRef] [PubMed] [Google Scholar]
© 2013 médecine/sciences – Inserm / SRMS
Liste des figures
|
Figure 1. Le double système de protection contre l’effet génotoxique des aldéhydes dans le contexte de la réparation de l’ADN. A. L’action enzymatique catabolique des aldéhydes déshydrogénases (Aldh2 ou Aldh) permet de détoxifier la cellule de ces composés très réactifs. Malgré cette première barrière, des quantités résiduelles d’aldéhydes (acétaldéhyde [CH3-CHO] ou autres aldéhydes [R-CHO]) peuvent induire des lésions de l’ADN. Ces lésions sont ensuite réparées par le système des protéines Fanconi. Ce mécanisme de protection à deux niveaux permet ainsi de maintenir l’intégrité du génome des cellules souches hématopoïétiques et la production contrôlée de cellules du sang. B. Dans le cas des souris Fancd2−/−Aldh2−/−, l’accumulation d’aldéhydes non oxydés a pour conséquence une augmentation du nombre de lésions de l’ADN dans les cellules souches hématopoïétiques. Le système de réparation par les protéines Fanconi étant incomplet et défectueux en raison de la mutation du gène Fancd2, ces lésions perdurent et aboutissent à l’émergence de cellules leucémiques ou à l’épuisement du stock de cellules souches hématopoïétiques. Ce modèle explique donc le développement de leucémie et d’anémie aplasique chez les souris Fancd2−/−Aldh2−/−. CSH : cellule souche hématopoïétique ; Aldh2 : aldéhyde déshydrogénase 2 ; Aldh : aldéhydes déshydrogénases ; CH3-CHO : acétaldéhyde ; R-CHO : aldéhydes. |
| Dans le texte | |
Les statistiques affichées correspondent au cumul d'une part des vues des résumés de l'article et d'autre part des vues et téléchargements de l'article plein-texte (PDF, Full-HTML, ePub... selon les formats disponibles) sur la platefome Vision4Press.
Les statistiques sont disponibles avec un délai de 48 à 96 heures et sont mises à jour quotidiennement en semaine.
Le chargement des statistiques peut être long.