")
")
| Issue |
Med Sci (Paris)
Volume 28, Number 6-7, Juin–Juillet 2012
|
|
|---|---|---|
| Page(s) | 612 - 617 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/2012286014 | |
| Published online | 16 juillet 2012 | |
Le stress dans tous ses états
Overview of acute and chronic stress responses
1
Institut national de la recherche agronomique (INRA), nutrition et neurobiologie intégrée, UMR 1286, 146, rue Léo Saignat, 33076 Bordeaux, France
2
Université de Bordeaux, nutrition et neurobiologie intégrée, UMR 1286, Bordeaux, France
3
Inserm U862, neurocentre Magendie, université de Bordeaux, Bordeaux, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
**
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Le concept psychobiologique du stress s’est construit progressivement depuis les années 1930. La compréhension des mécanismes neurobiologiques sous-jacents a connu une évolution spectaculaire ces dernières années. Cette synthèse fait le point sur ces connaissances en opposant stress aigu et stress chronique. Le premier correspond à une réponse adaptative et nécessaire à la survie. Il met en jeu de multiples médiateurs, neurotransmetteurs, neuropeptides et hormones, coordonnés dans le temps et l’espace. Le stress chronique est, en revanche, délétère et conduit, chez l’individu vulnérable, à diverses pathologies. Ses effets néfastes résultent d’une dérégulation des systèmes de stress dont les divers mécanismes évoqués sont présentés dans cet article.
Abstract
The psychobiological concept of stress built up over the years since the 1930s. The understanding of the neurobiological mechanisms involved has progressed remarkably during the past years. This article provides an overview of the recent data opposing acute and chronic stress. The former is an adaptative response of the organism to cope with the fluctuations of the environment and thereby is essential for survival. By contrast, chronic stress is deleterious and leads to various disease states in vulnerable individuals. Its adverse effects on the brain and the body result from a dysregulation of the stress system with various origins and mechanisms which we will discuss in this review.
© 2012 médecine/sciences – Inserm / SRMS
Depuis quelques décennies le terme de stress est passé dans le langage commun et est invoqué comme facteur ou processus conduisant, ou du moins contribuant, à des pathologies aussi diverses que les maladies cardiovasculaires, les troubles de l’humeur et l’anxiété, les désordres métaboliques, les pathologies auto-immunes et inflammatoires et les troubles musculosquelettiques. Nous sommes tous concernés par le stress qui se manifeste dans notre vie sociale, affective mais aussi professionnelle. D’un point de vue scientifique, depuis la première définition de Hans Selye en 1936, les concepts associés au stress ont largement évolué, passant d’une réaction physiologique non spécifique à des processus psychobiologiques complexes fortement dépendants de l’individu. Les réponses de stress sont nécessaires à la survie et ne deviennent délétères que lorsqu’elles sont sollicitées de façon chronique. Cette synthèse a pour objectif d’exposer et de discuter l’état des connaissances sur la psychobiologie du stress.
Stresseurs et stress
Le terme stress et certains aspects du concept qui lui est rattaché ont été introduits par Hans Selye en 1936. Ce concept est contemporain de la notion d’homéostasie proposée par Walter Cannon en continuité avec celle de « fixité du milieu intérieur » développée auparavant par Claude Bernard vers 1850. Stress, mot anglais qui vient de la mécanique et veut dire contrainte, charge, est utilisé d’une manière inconsidérée, et le flou lexical et sémantique est dû à H. Selye lui-même. En toute rigueur, « stress » est le « stresseur » (facteur de stress), et « strain » - qui se traduit par surmenage et fatigue - le processus qui survient en réponse au stress. Il faut donc distinguer, pour le moins, « stresseur » et « stress » si ce dernier garde - à tort - le sens que lui a donné Selye : la conséquence biologique [1, 2].
Selon Hans Selye, le stress est la « réponse de l’organisme à toute sollicitation qui lui est faite ». Il se caractérise par une réaction physiologique linéaire (libération de cortisol) qui n’est pas spécifique du stresseur. Ce concept a commencé à être contredit à partir des années 1960, par exemple par John Mason [3] qui a démontré l’importance de l’activation émotionnelle dans l’intensité des réponses de stress. Par la suite, la théorie cognitive du stress fut élaborée par plusieurs auteurs : par exemple Seymour Levine montra, dans une série d’études sur l’aversion conditionnée au goût chez l’animal, les influences de la nouveauté, de l’imprévisibilité et du manque de contrôle sur la modulation de la réponse biologique de stress [4]. L’évaluation cognitive est cruciale dans l’activation émotionnelle et physiologique qui s’ensuit, ce qui rejoint les théories de coping élaborées par les neuropsychologues. Ainsi, Robert Lazarus et Susan Folkman [5] ont énoncé le concept transactionnel du stress défini comme « le déséquilibre entre les sollicitations faites à l’individu et les ressources dont il dispose pour les affronter ». Cette théorie prend en compte la forte variabilité interindividuelle observée dans les réponses de stress, relevant à la fois du patrimoine génétique et de l’histoire personnelle. Depuis, les progrès de la neurobiologie, par le biais d’études chez des modèles animaux et de la neuroimagerie chez l’homme, ont permis de décrire un ensemble complexe d’événements neurobiologiques mis en jeu lorsque l’individu est soumis à un stresseur.
Ainsi, le stress peut être considéré comme un concept fondamentalement psychobiologique, les stresseurs agissant par l’intermédiaire de processus cognitifs et émotionnels, tout phénomène mental ayant par essence une correspondance cérébrale et biologique.
Le stress aigu est adaptatif
De multiples médiateurs biologiques sont impliqués
Un événement isolé, ressenti comme une menace, une situation d’incertitude ou d’imprévisibilité, conduisent à un stress aigu et à la recherche d’un nouvel équilibre. La mise en place des réponses psychobiologiques est généralement inconsciente (Figure 1).
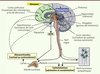 |
Figure 1. Médiateurs biologiques des réponses de stress. Le cerveau occupe une place centrale dans les réponses de stress aigu et permet l’action concertée de l’ensemble des médiateurs biologiques impliqués à travers des réseaux neuronaux interconnectés. Selon les espèces, le glucocorticoïde majeur est le cortisol (homme) ou la corticostérone (rongeurs) (d’après [25]). |
L’amygdale (ou complexe amygdalien : ensemble de noyaux situé dans le lobe temporal médian, une région impliquée dans les intégrations émotionnelles), en interaction avec le locus cœruleus (noyau du tronc cérébral recevant les informations de la périphérie) a un rôle clé dans l’orchestration des réponses comportementales et biologiques de stress dans le cerveau et le reste du corps. Ces deux structures sont interconnectées par leurs projections neuronales réciproques et entraînent une prompte activation du cerveau. Après une première phase de perception inconsciente, l’activation de l’amygdale est, dans une deuxième phase, modulée par ses connexions neuronales, d’une part avec le cortex préfrontal - région déterminante pour l’évaluation cognitive des informations reçues - et, d’autre part, avec l’hippocampe - structure clé dans la mémoire des expériences similaires déjà vécues. L’intervention de ces structures cérébrales contribue largement à la variabilité individuelle observée dans les réponses de stress [6].
La stimulation du locus cœruleus active en particulier le cortex préfrontal par le biais de ses nombreuses projections noradrénergiques. La stimulation de l’amygdale entraîne la libération rapide de neurotransmetteurs tels que la dopamine, l’acétylcholine, la sérotonine et la noradrénaline, et de peptides tels que la corticolibérine (appelée CRH pour corticotropin releasing hormone) dont les cellules productrices sont interconnectées avec le locus cœruleus. Ces facteurs permettent un éveil émotionnel, une augmentation de la vigilance et du traitement des informations reçues, et conduisent à un choix de la stratégie optimale pour faire face au stresseur [7, 8].
La réponse biologique de stress se propage dans le reste du corps par l’action de la CRH. En agissant sur le locus cœruleus, la CRH conduit à la synthèse et la libération d’adrénaline et de noradrénaline par la medulla des glandes surrénales, ainsi qu’à la libération de noradrénaline par les nerfs terminaux sympathiques dans tout le corps. Parallèlement, la stimulation de l’amygdale active l’axe corticotrope en provoquant la synthèse de CRH dans l’hypothalamus. La CRH et d’autres sécrétagogues dont l’arginine vasopressine (AVP) vont être transportés de l’hypothalamus vers l’hypophyse via le système porte et conduire à la libération d’adrénocorticotrophine (ACTH) dans le sang. À son tour, l’ACTH va stimuler la synthèse et la libération des hormones glucocorticoïdes (cortisol chez l’homme, corticostérone chez les rongeurs de laboratoire) à partir du cortex des glandes surrénales.
En périphérie, les hormones du stress (glucocorticoïdes et catécholamines) vont agir en augmentant le tonus vasculaire, la pression artérielle et la fréquence respiratoire. Ces mêmes hormones vont mobiliser les facteurs énergétiques (par leur action catabolique sur les protéines et les lipides) et les diriger vers les muscles et le cerveau afin de subvenir aux besoins des réponses comportementales (par exemple la fuite ou le combat). Enfin, les hormones du stress vont transitoirement accroître l’immunité et inhiber, également transitoirement, des fonctions coûteuses en énergie telles que la digestion, la croissance et la reproduction.
Ces modifications biologiques doivent être limitées dans le temps pour ne pas affecter l’organisme. Les hormones du stress, cortisol et catécholamines, vont agir en retour sur le cerveau pour éteindre les réponses de stress et recouvrer l’homéostasie, ainsi que pour stocker dans la mémoire les nouvelles informations qui seront utiles lors d’une future exposition à un stresseur similaire.
Les réponses de stress sont organisées dans le temps et l’espace
Les différentes molécules impliquées ont chacune des niches spatiotemporelles spécifiques, conditionnées par les sites de libération de ces médiateurs, la localisation spécifique de leurs récepteurs ainsi que l’affinité et le type de molécules réceptrices. Typiquement, les neurotransmetteurs (noradrénaline, sérotonine, dopamine) et les peptides (CRH, AVP) agissent très vite, dans les minutes - voire les secondes - qui suivent l’apparition du stresseur, et cessent leur activité tout aussi rapidement. Ils exercent leurs actions via des récepteurs membranaires couplés aux protéines G qui transfèrent rapidement leur activation à des effecteurs en aval, altérant ainsi le fonctionnement des neurones exprimant ces récepteurs.
En revanche, l’action des hormones glucocorticoïdes est plus tardive. Ces hormones se lient à des récepteurs intracellulaires (récepteurs aux minéralocorticoïdes [MR] et aux glucocorticoïdes [GR]) qui migrent ensuite vers le noyau où ils agissent en tant que facteurs de transcription en altérant l’expression de gènes, et donc certaines fonctions cellulaires. Ainsi, ces hormones agissent avec un délai d’environ une heure, mais leur action perdure plusieurs heures après l’apparition du stresseur. Ces réponses en vagues successives servent des fonctions différentes : la première vague promeut la vigilance, l’évaluation de la situation et la prise de décision, alors que la deuxième vague permet une réponse adaptative prolongée, par exemple la consolidation de la mémoire des informations liées au stresseur [9, 10] (Figure 2).
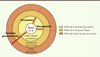 |
Figure 2. Décours temporel des réponses de stress. Les flèches épaisses montrent la durée d’action des différents médiateurs qui est liée aux types de récepteurs mis en jeu. Cependant ce découpage temporel n’est pas aussi strict, comme schématisé par les flèches fines. Les hormones glucocorticoïdes ont aussi des actions rapides via leur liaison à des récepteurs membranaires. Les neuropeptides et monoamines entraînent des changements génomiques prolongés en régulant des facteurs de transcription (adapté de [10]). |
Enfin, l’action concertée de toutes ces molécules médiatrices du stress est rendue possible par la convergence spatiale de leurs actions sous forme de réseaux, les « points chauds » étant le cortex préfrontal, l’amygdale, l’hippocampe et les lieux de synthèse des neurotransmetteurs (le locus cœruleus et les noyaux du raphé). Bien que chaque médiateur possède des caractéristiques propres, sa contribution fonctionnelle à la réponse de stress dépend de son interaction avec les autres médiateurs par le biais de ces réseaux interconnectés.
Le terme de « neurosymphonie du stress » a été employé par M. Joëls et T.Z. Baram [10] pour qualifier ce système complexe de réponses de stress, si finement coordonné, très conservé au cours de l’évolution et surtout essentiel à la survie.
Le stress chronique est une désadaptation conduisant à la pathologie
Dans des situations de stress chroniques ou intenses, la permanence des stresseurs empêche la récupération et le retour à l’équilibre, et des traces perdurent dans l’organisme, objectivées par des symptômes biologiques et psychologiques. Il est ici question du « passage du normal au pathologique ». Les changements pathologiques se révèlent dans les jours, les semaines, voire les années qui suivent les événements stressants par une dérégulation des systèmes de stress.
L’excès d’hormones glucocorticoïdes est néfaste
Le stress chronique est classiquement associé à une sécrétion excessive de cortisol. R. M. Sapolsky a été le premier à évoquer l’hypothèse de la cascade glucocorticoïde pour expliquer l’altération des fonctions cérébrales due à une exposition prolongée aux glucocorticoïdes [11] (Figure 3). Selon ce concept, la sécrétion répétée ou prolongée d’hormone glucocorticoïde endommage en particulier les neurones hippocampiques (riches en récepteurs de cette hormone) et conduit à une levée de l’inhibition tonique qu’exerce l’hippocampe sur l’activation de l’axe corticotrope. La surproduction de glucocorticoïdes se pérennise via un cercle vicieux qui conduit à divers processus pathologiques (Figure 3).
Le postulat d’un effet direct des glucocorticoïdes sur le processus de neurodégénération est très controversé. En effet, chez les patients souffrant d’un syndrome de Cushing (tumeur de l’hypophyse ou des surrénales entraînant une sécrétion élevée et continue de cortisol), on observe une réduction du volume de l’hippocampe qui est en partie réversible après quelques semaines de traitement, suggérant qu’il n’y a pas de perte neuronale totale chez ces patients. Selon l’hypothèse actuelle, les glucocorticoïdes favoriseraient la neurotoxicité exercée par les acides aminés excitateurs (l’excitotoxicité) [9, 12, 13]. Cette excitotoxicité se traduit par des changements morphologiques de certaines régions du cerveau qui modifient les connectivités synaptiques et donc le fonctionnement cérébral. Dans l’hippocampe, la diminution de l’arborisation des neurones de la région CA3 et une rétraction de leurs dendrites ainsi qu’une perte des épines dendritiques après un stress prolongé, diminuent de fait le nombre de connexions synaptiques entre neurones. Ce phénomène a également été observé dans le cortex préfrontal médian, alors que dans le noyau basolatéral de l’amygdale et le cortex orbitofrontal, on observe une hypertrophie des neurones. Parallèlement, la neurogenèse est ralentie dans le gyrus denté (hippocampe) et la survie des progéniteurs des neurones et des neurones matures de cette région de l’hippocampe est affectée.
Le glutamate, régulé par les glucocorticoïdes, semble être causal dans les altérations structurales de l’hippocampe puisque le blocage pharmacologique de ses récepteurs prévient l’atrophie dendritique observée dans les situations de stress chronique. Dans le noyau basolatéral de l’amygdale, l’inhibition de l’acide ³-aminobutyrique (GABA) par les glucocorticoïdes serait responsable de la croissance dendritique et du développement des épines [7].
Fonctionnellement, ces changements morphologiques dans le cerveau sont associés dans le cortex préfrontal à une altération de l’attention, de la mémoire de travail et de la flexibilité comportementale. Les modifications structurales dans l’hippocampe sont associées à des troubles de la mémoire spatiale et de l’apprentissage alors que dans l’amygdale, l’hypertrophie dendritique est associée à une augmentation de la mémoire de peur, à de l’anxiété et de l’agressivité de la part des animaux. Quelques mois après l’arrêt des stresseurs, les neurones de l’hippocampe et du cortex préfrontal médian reviennent à leur conformation d’origine, ce qui confirme la plasticité neuronale. En revanche, l’hypertrophie de l’amygdale persiste après l’arrêt des stresseurs dans les modèles utilisés jusqu’à présent [7] (Figure 4).
 |
Figure 4. Modifications structurales et fonctionnelles dans des régions interconnectées du cerveau suite à un stress chronique. GR : récepteur des glucocorticoïdes ; CRH : corticotrophin releasing hormone ou corticolibérine (adapté de [7]). |
Le rôle du remodelage des dendrites est considéré comme un processus adaptatif par certains auteurs : il éviterait une suractivité du glutamate qui engendrerait des effets neurotoxiques irréversibles. Ce phénomène est d’ailleurs observé lors d’un stress aigu quoique de manière moins développée qu’en réponse à un stress chronique [12]. D’autres chercheurs pensent, au contraire, que la réduction de la taille et de la densité des épines dendritiques accroît la vulnérabilité des neurones aux effets neurotoxiques irréversibles [14].
L’hypoactivité des glucocorticoïdes est aussi délétère
À l’inverse de l’hypothèse de la cascade glucocorticoïde, un ensemble de données fait apparaître qu'une déficience du signal glucocorticoïde peut engendrer des effets aussi délétères que l'hyperactivité de cette hormone [15]. On l’observe en effet dans plusieurs maladies dites liées au stress comme le syndrome de fatigue chronique, la fibromyalgie, l’épuisement professionnel ou l’état de stress post-traumatique [15–17]. De plus, une série d’études récentes montrent que le stress chronique chez l’homme est associé à une réduction de l’action du cortisol qui favoriserait l’activité pro-inflammatoire [18]. Plusieurs mécanismes peuvent conduire à une diminution de l’efficacité des glucocorticoïdes : une production et/ou une biodisponibilité réduites (appelée hypocortisolisme), une désensibilisation des récepteurs d’un sécrétagogue (CRH, AVP ou ACTH), une sensibilité augmentée au rétrocontrôle négatif ou une réponse réduite des tissus cibles, qu’entraînerait par exemple une désensibilisation des récepteurs au cortisol (MR ou GR). Les mécanismes moléculaires conduisant à une désensibilisation ou à une hypersensibilisation de récepteurs sont probablement d’origine épigénétique. Par exemple, l’absence de soins maternels chez le raton entraîne une hyperméthylation du gène codant pour le récepteur GR dans l’hippocampe, réduisant ainsi le nombre de récepteurs dans cette structure et, par conséquent, l’efficacité du rétrocontrôle négatif de la sécrétion des glucocorticoïdes [19]. Le même mécanisme est évoqué chez l’homme en réponse à la maltraitance chez l’enfant, qui conduit à des comportements dépressifs et suicidaires [20]. Le test de freinage à la dexaméthasone1, qui permet d’évaluer l’efficacité du rétrocontrôle des glucocorticoïdes, est d’ailleurs utilisé comme marqueur des états dépressifs. Une revue récente conclut qu’un hypocortisolisme se développe au décours d’un stress chronique prolongé, ou peut succéder à un état initial d’hyperactivité corticotrope qui évoluerait finalement en une hypoactivité corticotrope [21].
Un défaut du signal glucocorticoïde a des conséquences physiopathologiques variées liées à l’excès d’activité des systèmes et des voies de signalisation qui sont normalement freinés par des niveaux efficaces de glucocorticoïdes. On peut mentionner la réponse immunitaire innée (source d’inflammation), l’activation du système nerveux sympathique et les voies de signalisation du CRH (Figure 5). L’inflammation, quand elle n’est pas maîtrisée, conduit à une augmentation de la libération de cytokines pro-inflammatoires qui peut participer à divers processus pathologiques dans l’organisme dont les maladies cardiovasculaires, le diabète, certains troubles psychiatriques voire le cancer [15, 22, 23].
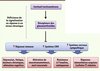 |
Figure 5. Hypothèse de la signalisation glucocorticoïde déficiente suite à un stress chronique. Les systèmes normalement inhibés par les glucocorticoïdes sont suractivés et induisent différentes pathologies (adapté de [15]). |
Conclusions
L’étude du stress et de ses conséquences physiopathologiques est un domaine de recherche très actif. Au Canada, c’est tout un institut qui y est consacré : le centre d’étude du stress humain de Montréal (www.stresshumain.ca). En France, un club stress rassemblant les neurobiologistes intéressés par le sujet, a récemment vu le jour au sein de la Société française des neurosciences.
Cette synthèse non exhaustive souligne la sophistication des mécanismes biologiques de stress mis en place pour que les individus s’adaptent aux fluctuations de leur environnement. Le paradoxe est que ces mécanismes ne semblent pas adaptés aux situations de stress chronique que nous subissons dans nos sociétés modernes, en particulier celles qu’engendre la complexité des rapports sociaux. En tout cas, une grande variabilité individuelle existe dans les réponses de stress et la susceptibilité aux maladies qui leurs sont associées. Cette variabilité, que nous n’avons pas abordée dans cette synthèse par manque de place, est en partie d’origine génétique, mais aussi due aux évènements de vie. Une sensibilité particulière au stress est observée au cours du développement, de la période périnatale jusqu’à l’adolescence, façonnant la personnalité psychobiologique des individus. Le sujet âgé, dont l’axe corticotrope s’est modifié au cours du temps, est également très vulnérable aux effets du stress [24].
Liens d’Intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
La dexaméthasone est un glucocorticoïde synthétique qui, lorsqu’il est administré chez un patient, freine l’axe corticotrope par rétrocontrole négatif. Chez les dépressifs on observe dans la majorité des cas un échappement à la dexaméthasone, c’est-à-dire que le freinage de la sécrétion des glucocorticoïdes le matin (< 50 ng/ml de cortisol) n’est pas obtenu pour une dose de 1 mg de dexaméthasone administrée la veille au soir.
Références
- Le Moal M. Historical approach, evolution of the stress concept: a personal account. Psychoneuroendocrinology 2007 ; 32 (suppl 1) : 3–9. [CrossRef] [Google Scholar]
- Dantzer R. Psychoneuroendocrinology of stress. In : Koob GF, Le Moal M, Thompson RF, eds. Encyclopedia of behavioral neurosciences, vol. 3. Oxford : Academic Press/Elsevier, 2010 : 126–131. [CrossRef] [Google Scholar]
- Mason JW. A re-evaluation of the concept of “non-specificity” in stress theory. J Psychiatr Res 1971 ; 8 : 323–333. [CrossRef] [PubMed] [Google Scholar]
- Hennessy JW, Smotherman WP, Levine S. Conditioned taste aversion and the pituitary-adrenal system. Behav Biol 1976 ; 16 : 413–424. [CrossRef] [PubMed] [Google Scholar]
- Lazarus RS. Coping theory and research: past, present, and future. Psychosom Med 1993 ; 55 : 234–247. [PubMed] [Google Scholar]
- Rodrigues SM, LeDoux JE, Sapolsky RM. The influence of stress hormones on fear circuitry. Annu Rev Neurosci 2009 ; 32 : 289–313. [CrossRef] [PubMed] [Google Scholar]
- Roozendaal B, McEwen BS, Chattarji S. Stress, memory and the amygdala. Nat Rev Neurosci 2009 ; 10 : 423–433. [CrossRef] [PubMed] [Google Scholar]
- Arnsten AF. Stress signalling pathways that impair prefrontal cortex structure and function. Nat Rev Neurosci 2009 ; 10 : 410–422. [CrossRef] [PubMed] [Google Scholar]
- De Kloet ER, Joels M, Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci 2005 ; 6 : 463–475. [CrossRef] [PubMed] [Google Scholar]
- Joels M, Baram TZ. The neuro-symphony of stress. Nat Rev Neurosci 2009 ; 10 : 459–466. [PubMed] [Google Scholar]
- Sapolsky RM, Krey LC, McEwen BS. The neuroendocrinology of stress and aging: the glucocorticoid cascade hypothesis. Endocr Rev 1986 ; 7 : 284–301. [CrossRef] [PubMed] [Google Scholar]
- McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev 2007 ; 87 : 873–904. [CrossRef] [PubMed] [Google Scholar]
- Groc L, Chaouloff F. Axe corticotrope et plasticité de la communication neuronale: décryptage des mécanismes cellulaires. Med Sci (Paris) 2008 ; 24 : 776–778. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Chaouloff F, Groc L. Temporal modulation of hippocampal excitatory transmission by corticosteroids and stress. Front Neuroendocrinol 2011 ; 32 : 25–42. [CrossRef] [PubMed] [Google Scholar]
- Raison CL, Miller AH. When not enough is too much: the role of insufficient glucocorticoid signaling in the pathophysiology of stress-related disorders. Am J Psychiatry 2003 ; 160 : 1554–1565. [CrossRef] [PubMed] [Google Scholar]
- Heim C, Ehlert U, Hellhammer DH. The potential role of hypocortisolism in the pathophysiology of stress-related bodily disorders. Psychoneuroendocrinology 2000 ; 25 : 1–35. [CrossRef] [PubMed] [Google Scholar]
- Yehuda R. Status of glucocorticoid alterations in post-traumatic stress disorder. Ann NY Acad Sci 2009 ; 1179 : 56–69. [CrossRef] [Google Scholar]
- Cole SW. Elevating the perspective on human stress genomics. Psychoneuroendocrinology 2010 ; 35 : 955–962. [CrossRef] [PubMed] [Google Scholar]
- Meaney MJ, Szyf M. Environmental programming of stress responses through DNA methylation: life at the interface between a dynamic environment and a fixed genome. Dialogues Clin Neurosci 2005 ; 7 : 103–123. [PubMed] [Google Scholar]
- McGowan PO, Sasaki A, D’Alessio AC, et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci 2009 ; 12 : 342–348. [CrossRef] [PubMed] [Google Scholar]
- Miller GE, Chen E, Zhou ES. If it goes up, must it come down? Chronic stress and the hypothalamic-pituitary-adrenocortical axis in humans. Psychol Bull 2007 ; 133 : 25–45. [CrossRef] [PubMed] [Google Scholar]
- Irwin MR, Cole SW. Reciprocal regulation of the neural and innate immune systems. Nat Rev Immunol 2011 ; 11 : 625–632. [CrossRef] [PubMed] [Google Scholar]
- Webster Marketon JI, Glaser R. Stress hormones and immune function. Cell Immunol 2008 ; 252 : 16–26. [CrossRef] [PubMed] [Google Scholar]
- Lupien SJ, McEwen BS, Gunnar MR, Heim C. Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat Rev Neurosci 2009 ; 10 : 434–445. [CrossRef] [PubMed] [Google Scholar]
- Inserm. Stress au travail et santé : situation chez les indépendants, vol. 483. Collection Expertise collective. Paris : Inserm, 2011. [Google Scholar]
Liste des figures
|
Figure 1. Médiateurs biologiques des réponses de stress. Le cerveau occupe une place centrale dans les réponses de stress aigu et permet l’action concertée de l’ensemble des médiateurs biologiques impliqués à travers des réseaux neuronaux interconnectés. Selon les espèces, le glucocorticoïde majeur est le cortisol (homme) ou la corticostérone (rongeurs) (d’après [25]). |
| Dans le texte | |
|
Figure 2. Décours temporel des réponses de stress. Les flèches épaisses montrent la durée d’action des différents médiateurs qui est liée aux types de récepteurs mis en jeu. Cependant ce découpage temporel n’est pas aussi strict, comme schématisé par les flèches fines. Les hormones glucocorticoïdes ont aussi des actions rapides via leur liaison à des récepteurs membranaires. Les neuropeptides et monoamines entraînent des changements génomiques prolongés en régulant des facteurs de transcription (adapté de [10]). |
| Dans le texte | |
 |
Figure 3. Hypothèse de la cascade glucocorticoïde ou hypothèse neurotoxique (adapté de [11]). |
| Dans le texte | |
|
Figure 4. Modifications structurales et fonctionnelles dans des régions interconnectées du cerveau suite à un stress chronique. GR : récepteur des glucocorticoïdes ; CRH : corticotrophin releasing hormone ou corticolibérine (adapté de [7]). |
| Dans le texte | |
|
Figure 5. Hypothèse de la signalisation glucocorticoïde déficiente suite à un stress chronique. Les systèmes normalement inhibés par les glucocorticoïdes sont suractivés et induisent différentes pathologies (adapté de [15]). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.