")
")
| Issue |
Med Sci (Paris)
Volume 27, Number 3, Mars 2011
|
|
|---|---|---|
| Page(s) | 297 - 302 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/2011273297 | |
| Published online | 30 March 2011 | |
Cellules souches mésenchymateuses
Production à usage clinique et contraintes sécuritaires
Mesenchymal stem cells
The challenge of a good therapeutic product
Établissement français du sang Pyrénées-Méditerranée, UMR5273 STROMALab-Inserm U1031, 75, rue de Lisieux, 31300 Toulouse, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
Les cellules souches mésenchymateuses ou cellules stromales mésenchymateuses multipotentes (CSM) appartiennent à une population cellulaire initialement identifiée dans la moelle osseuse mais présente dans tous les tissus. Par leur potentiel de différenciation, leur production de cytokines, de facteurs trophiques et leurs actions immunosuppressives, les CSM sont un outil thérapeutique tant en médecine régénérative que dans le traitement des pathologies immunitaires et inflammatoires. Actuellement, une centaine d’essais utilisant des CSM sont officiellement répertoriés. Un prérequis pour l’utilisation thérapeutique des CSM est leur conformité avec les standards de « bonnes pratiques de fabrication » (BPF ou good manufacturing practices, GMP) assortis de contrôles de sécurité adéquats. Le défi est de passer de procédés de culture utilisés en recherche à des procédés de production correspondant à ces standards et à la réglementation nationale et internationale (européenne et nord-américaine). Ceci nécessite un travail de recherche et développement en liaison directe avec les équipes de recherche et les équipes cliniques qui mettent en place les essais cliniques. C’est cette intégration verticale, assurant des allers-retours permanents entre la recherche, le développement et la clinique, qui permettra les développements pertinents débouchant sur les procédés et les indications définitifs.
Abstract
Mesenchymal stem cells (or stromal cells) have been initially characterized in bone marrow, but since, they have been identified in almost every tissue. Their multiple properties, namely differentiative capacity, production of cytokines and trophic molecules, and their immunosuppressive potential undoubtedly offer many therapeutic advantages, both for regenerative medecine or to relieve immune or inflammatory diseases. This is illustrated by the high number (> 100) of ongoing clinical trials with these cells. However, a prerequsite for their safe use in clinics is to guarantee that their production meet the good manufacturing practices, and that the final product is validated by adequate controls. It is thus quite a challenge to move from procedures defined for a research use to large scale production that fits with the national and international rules in terms of standardisation and controls. This underlines the importance of developping interacting networks between research teams, physicians and the industrial R&D departments. This fruitful collaboration will ensure the definition of appropriate and safe procedures for a successful therapeutic application.
© 2011 médecine/sciences - Inserm / SRMS
Cellules souches mésenchymateuses : quelle réglementation ?
En Europe, les CSM sont considérées comme des médicaments de thérapie innovante (MTI ou advanced therapy medicinal products, ATMP) comme le définit la directive (CE) no 1394/2007 du Parlement européen et du Conseil1. Selon cette directive, les CSM sont considérées soit comme des produits thérapeutiques qui proviennent de cellules somatiques, soit comme des produits d’ingénierie tissulaire, selon leur origine, leur procédé de production et les indications thérapeutiques (Encadré 1). Cette réglementation décrit les procédures d’autorisation et les exigences techniques concernant les caractéristiques du produit final et les règles d’étiquetage et d’emballage, que ces produits soient issus de l’industrie ou d’institutions académiques. De plus, cette réglementation se réfère aux règles européennes des BPF. Même si les CSM sont produites dans des laboratoires de recherche depuis plus de 40 ans [1, 2], le passage des protocoles de recherche à des protocoles de production à grande échelle respectant les BPF nécessite une analyse approfondie de tous les aspects critiques [3]. Comme pour toute production de cellules à usage clinique, la production des CSM doit se faire dans un environnement spécifique, sous une hotte à flux laminaire (classe A) placée dans une pièce qui est en surpression et dont on maintient le taux d’empoussièrement très bas (classe B). De même, tous les réactifs et les substances utilisés dans le procédé de production et les contrôles, le stockage, la réception, l’envoi et la distribution doivent respecter cette réglementation. Ces exigences demeurent tant qu’il persiste des étapes ouvertes au cours de la production.
La production des CSM à usage clinique
De nombreux paramètres doivent être pris en compte pour la production de CSM à usage clinique. Deux éléments sont essentiels : le choix du matériel de départ et les procédés d’obtention de CSM de « grade clinique ».
Règlement (CE) n° 1394/2007 du Parlement européen et du Conseil du 13 novembre 2007 concernant les médicaments de thérapie innovante et modifiant la directive 2001/83/CE ainsi que le règlement (CE) n° 726/2004
-
Des progrès scientifiques récents en biotechnologie cellulaire et moléculaire ont conduit à la mise au point de thérapies innovantes, telles que la thérapie génique, la thérapie cellulaire somatique ou l’ingénierie tissulaire. Cette discipline naissante, la biomédecine, offre de nouvelles possibilités de traitement des maladies et des dysfonctionnements du corps humain.
-
Dans la mesure où les produits de thérapie innovante sont présentés comme ayant des propriétés curatives ou préventives à l’égard des maladies humaines, ou comme pouvant être utilisés chez l’homme ou administrés à celui-ci en vue de restaurer, corriger ou modifier des fonctions physiologiques en exerçant une action principalement pharmacologique, immunologique ou métabolique, ils constituent des médicaments biologiques au sens de l’annexe I de la directive 2001/83/CE du Parlement européen et du Conseil du 6 novembre 2001 instituant un code communautaire relatif aux médicaments à usage humain.
…
Outre les définitions figurant à l’article 1er de la directive 2001/83/CE et à l’article 3, points à) à 1) et 0) à q), de la directive 2004/23/CE, les définitions suivantes s’appliquent aux fins du présent règlement :
-
« médicament de thérapie innovante » : l’un des médicaments à usage humain suivants :
un médicament de thérapie génique tel que défini dans l’annexe I, partie IV, de la directive 2001/83/CE,
un médicament de thérapie cellulaire somatique tel que défini dans l’annexe I, partie IV, de la directive 2001/83/CE,
un produit issu de l’ingénierie tissulaire tel que défini au point b) ;
-
« produit issu de l’ingénierie tissulaire » :
un produit qui contient des cellules ou tissus issus de l’ingénierie cellulaire ou tissulaire, ou en est constitué, et
qui est présenté comme possédant des propriétés lui permettant de régénérer, réparer ou remplacer un tissu humain, ou est utilisé chez l’être humain ou administré à celui-ci dans ce but.
Un produit issu de l’ingénierie tissulaire peut contenir des cellules ou des tissus d’origine humaine ou d’origine animale, ou les deux. Les cellules ou tissus peuvent être viables ou non viables. Il peut également contenir des substances supplémentaires, telles que des produits cellulaires, des biomolécules, des biomatériaux, des substances chimiques, des supports ou des matrices.
Le matériel de départ
Un paramètre reste sous-estimé : le donneur lui-même. Actuellement, comme pour tous les produits cellulaires ou tissulaires, la sélection des donneurs repose essentiellement sur l’âge (les donneurs sont a priori des adultes jeunes) et sur l’absence de marqueurs positifs pour les agents infectieux transmissibles. Bien qu’indispensables, ces paramètres ne sont pas suffisants pour identifier les « bons » donneurs pour la production de CSM. Il est d’autant plus important de développer des critères de sélection stricts que certains préconisent la sélection d’un donneur unique pour la production de lots importants utilisables chez de très nombreux receveurs. L’âge du donneur est certainement important. Ainsi, en comparant le nombre de colony-forming unit fibroblast (CFU-F) dans la moelle osseuse en fonction de l’âge des donneurs, on a pu montrer que la moelle des enfants est plus riche en CFU-F [4]. De plus, une perte des capacités proliférative et de multipotentialité des CSM peut être directement liée à l’âge du donneur [5]. De fait, il faudrait pouvoir s’assurer que le donneur n’a pas d’anomalie ou de risque d’anomalies pouvant toucher les CSM, ce qui est difficile. Si, en ce domaine, il n’existe pas d’exigence de la part des autorités de régulation, il faudra se pencher sur ce problème, surtout en cas d’utilisation de donneurs dits uniques.
De nombreux tissus sont des sources potentielles de CSM : la moelle osseuse, le tissu adipeux et le sang de cordon sont les plus utilisés actuellement [6]. La moelle osseuse et le tissu adipeux sont d’accès facile, et les CSM - dont la quantité est estimée par le nombre de CFU-F mesuré en culture - sont plus abondantes dans le tissu adipeux que dans la moelle osseuse [35] (→).
(→) Voir l’article de P. Charbord et L. Casteilla, page 261 de ce numéro
Les CSM sont rares dans le sang de cordon et le succès de leur obtention dépend étroitement du temps écoulé entre la délivrance et le recueil du sang de cordon, du volume de sang et de la quantité de cellules recueillis [7]. Les CSM peuvent également être d’origine fœtale ou néonatale ; la membrane amniotique et la gelée de Wharton (conjonctif du cordon) contiennent de grandes quantités de CSM dont l’intérêt est la plus grande « souchitude » ou immaturité de ces cellules.
Les procédés
Densité d’ensemencement : un paramètre crucial
Les procédés de culture sont le deuxième paramètre majeur à prendre en considération. Les cellules mises en culture peuvent être soit les cellules totales extraites du tissu, soit une population particulière. À partir de la moelle osseuse, l’ensemencement peut se faire directement, sans étape préalable. Cependant, les CSM sont présentes au sein des cellules mononucléées de la moelle osseuse obtenues par centrifugation sur gradient de densité. Le tissu adipeux, quant à lui, doit d’abord être dissocié par digestion enzymatique, ce qui permet d’obtenir la fraction stromale vasculaire qui contient les CSM. Le matériel initial médullaire peut encore être enrichi en CSM par une seconde étape de sélection soit par cytométrie en flux, soit par la méthode immunomagnétique. Différents antigènes permettent en effet d’isoler des sous-populations riches en CSM. Ainsi, le tri par cytométrie en flux de la population STRO-1fort permet d’enrichir (x 950 fois) en CFU-F la suspension de cellules mononucléées et les cellules coexprimant STRO-1 et CD106 (vascular cell adhesion molecule, VCAM) représentent une population très purifiée de CSM [8]. Mais d’autres antigènes peuvent être utilisés avec des résultats performants : le CD49a ou le CD271 [9]. Le CD49a (ou chaîne alpha 1 des intégrines) est exprimé par les CSM et toutes les CFU-F sont présentes dans la fraction CD49a+ des cellules médullaires [10]. Ces procédures permettent un enrichissement en CSM, mais non l’obtention d’une population pure de CSM. Quelle que soit la source de cellules et la population cellulaire mise en culture, les CSM présentes dans le matériel mis en culture sont de toutes façons isolées par leur propriété d’adhérence à la surface du flacon de culture (le plus souvent du plastique). Les CSM étant des cellules adhérentes caractérisées par une inhibition de contact lorsqu’elles arrivent à confluence, la densité d’ensemencement est fondamentale pour l’obtention d’un taux d’expansion adéquat et le maintien des fonctions des CSM. Les densités d’ensemencement sont très variables, elles vont de 50 × 103 à 170 × 103 cellules/cm2. Après la première phase de culture, les cellules doivent être détachées et réensemencées dans un nouveau flacon (passage) et la densité d’ensemencement doit être fortement diminuée [11]. Le choix de la densité cellulaire d’ensemencement est un paramètre crucial ; l’utilisation d’une faible voire très faible densité d’ensemencement pourrait permettre de maintenir un fort taux d’expansion et la multipotentialité des CSM cultivées [12]. Pour la production à usage clinique, l’utilisation d’une densité très faible est difficile car elle nécessite de très grandes surfaces de culture. Ainsi, à partir du premier passage, une densité d’ensemencement de 1 000 cellules/cm2 semble être un bon compromis [13]. L’inhibition de contact des CSM à confluence, qui entraîne un arrêt de prolifération, conduit à réaliser des passages successifs pour obtenir de grandes quantités de CSM pures. C’est aussi nécessaire pour éliminer les possibles cellules hématopoïétiques - présentes dans l’inoculum initial de cellules médullaires - qui peuvent persister à la fin de la première phase de culture. Cependant, au cours de ces passages successifs, le taux de prolifération diminue, les cellules perdent progressivement leur multipotentialité et finalement ne sont plus capables que de se différencier en ostéoblastes [14, 15]. Cette perte de potentiel est pour partie liée au processus de sénescence qui apparaît en culture (voir le paragraphe sur les contrôles) dès 12 à 15 doublements de population (DP). En conséquence, pour des raisons tant d’efficacité que de sécurité, il semble opportun de limiter le nombre de DP en culture et de ne pas dépasser 20 DP.
Milieux de culture
Comme la densité cellulaire et le temps de culture, la composition du milieu est essentielle pour l’efficacité et la sécurité de la culture ex vivo des CSM. Les milieux doivent idéalement maintenir le phénotype, la stabilité génétique et les fonctions des CSM au cours des passages. De nombreux facteurs de croissance et cytokines sont efficaces sans que l’on connaisse avec certitude les besoins exacts pour les CSM, et ceux-ci dépendent des résultats attendus, par exemple du nombre de cellules désiré, du type et du stade de différenciation. Ceci étant, certains facteurs de croissance et cytokines sont cruciaux : le platelet-derived growth factor (PDGF), l’epidermal growth factor (EGF), le transforming growth factor β (TGF-β), l’insulin-like growth factor (IGF) et le fibroblast growth factor 2 (FGF2) [16]. Ainsi, le FGF2 permet une forte amplification du nombre de cellules tout en conservant mieux leur multipotentialité. Même s’il n’y a pas de consensus sur le milieu idéal pour la culture des CSM, le DMEM et l’αMEM2 sont les plus communément utilisés avec, en complément, soit du sérum de veau fœtal (SVF) utilisé à une concentration de 10 %, soit du plasma humain [17, 18], auxquels des facteurs de croissance sont ajoutés. L’utilisation du SVF fait débat : en effet, au-delà des risques infectieux, éliminés par un criblage attentif des lots de SVF utilisés au cours de la culture, les CSM peuvent retenir dans leur cytoplasme des protéines bovines qui pourraient provoquer des risques de sensibilisation. Certaines autorités de régulation, tel le PEI3 en Allemagne, s’apprêtent à interdire l’utilisation de SVF ; celui-ci est remplacé par des produits plus sécurisés d’origine humaine voire des milieux sans sérum complètement définis. Parmi les produits d’origine humaine provenant de sources sécurisées par le système transfusionnel, le sérum de groupe sanguin AB et le lysat plaquettaire ont été testés. Le plus simple et efficace est le lysat plaquettaire : il s’agit de plasma humain enrichi en facteurs de croissance d’origine plaquettaire. Pour ce faire, les concentrés plaquettaires provenant des centres de transfusion sont mélangés puis soumis à des cycles de congélation/décongélation provoquant la rupture des plaquettes et le relargage dans le plasma des facteurs de croissance qu’elles contiennent. Après filtration, le lysat plaquettaire peut être utilisé frais ou conservé congelé. De nombreuses équipes ont démontré l’efficacité au moins équivalente, voire supérieure, du lysat plaquettaire par rapport au SVF [17, 18]. Différents milieux sans sérum qui utilisent des combinaisons de facteurs de croissance (par exemple : FGF2, PDGF, TGF-β) ont été développés dans le cadre de la recherche. Ces milieux permettent d’obtenir des CSM dont le nombre et la qualité sont comparables à ceux obtenus par les procédés de culture classiques [19]. Cependant, il est nécessaire d’ajouter une protéine comme la fibronectine qui assure l’adhérence initiale des cellules. Aussi n’y a-t-il pas encore de milieu sans sérum répondant aux critères des BPF.
Sécurité sanitaire
Au-delà des différents paramètres et procédés évoqués, il est nécessaire d’assurer l’aseptie des conditions de culture (les règles en sont définies dans l’annexe 1 de la réglementation européenne des GMP4). Ceci conduit à privilégier des systèmes de culture clos par rapport aux systèmes classiques en flacons de culture. L’utilisation de containers multi-étages (de marque Nunc ou Corning) qui peuvent être couplés à des systèmes de connexion, tubulure et poches (Macopharma) est une première étape vers les systèmes totalement clos. Ce type de système permet d’obtenir de façon simple et sécurisée de grandes quantités de CSM, de quelques centaines de millions à plus d’un milliard [20]. Le passage de conditions de culture 2D à des conditions 3D peut accroître la fonctionnalité des CSM comme cela a été montré pour leurs propriétés anti-inflammatoires. Cependant, pour répondre complètement aux exigences des BPF, il est nécessaire d’utiliser un bioréacteur clos et automatisé. Les critères principaux pour ces bioréacteurs sont un rapport surface/volume élevé, un système clos et une automatisation de la mise en culture et du recueil des cellules. Différentes configurations de bioréacteurs peuvent permettre une amélioration de ces critères, que ce soit des plateaux multiples, des fibres creuses ou des systèmes microfluidiques [21]. Différentes compagnies comme Caridian BCT ont développé des bioréacteurs totalement automatisés qui permettent d’obtenir de grandes quantités de CSM en conditions BPF. Utilisé avec des milieux dépourvus de produits animaux voire totalement de sérum, ce type de technologie permet de produire des CSM en parfaite adéquation avec les contraintes des BPF.
Le contrôle des CSM cultivées
La sécurité de l’utilisation clinique des CSM est subordonnée aux contrôles réalisés lors du développement des procédés et lors de la libération des lots avant utilisation. Au cours du processus d’expansion ex vivo, les CSM sont exposées, comme toute cellule cultivée, à différents risques, dont en particulier : la contamination bactériologique ou virale, la contamination par des agents xénogéniques et l’instabilité génétique. On peut distinguer deux types de contrôles : d’une part, les contrôles mis en place lors du développement des procédés. Ils permettent la qualification des procédés et démontrent l’efficacité des CSM obtenues. D’autre part, les contrôles requis pour l’utilisation clinique des CSM produites. Ils assurent la sécurité et l’efficacité des CSM tout en permettant une libération rapide de ces dernières. Quels que soient les types de contrôles, ceux-ci doivent être standardisés. Les contrôles de libération des CSM comprennent des tests bactériologiques, des contrôles phénotypiques, et, si possible, des analyses fonctionnelles comme la mesure du nombre de CFU-F et de la sécrétion de certaines molécules. Cependant, ces contrôles ne peuvent pas être réalisés rapidement et, hormis une utilisation décalée dans le temps (après congélation des CSM), les résultats ne seront obtenus que a posteriori. Les contrôles finaux avant utilisation doivent inclure une évaluation de la viabilité et du phénotype.
La stabilité génétique des CSM
Une question centrale si l’on envisage une utilisation clinique des CSM est celle de la stabilité génétique des CSM en culture et des risques liés à la transformation potentielle des cellules en culture ou à l’utilisation de cellules sénescentes. En utilisant des CSM immortalisées après transduction de hTERT (human telomerase reverse transcriptase), on a pu montrer que la transformation des CSM est un processus long et multi-étapes qui fait intervenir la délétion de p16ink4a [22]. Depuis 2005, différents articles avaient rapporté que les CSM en culture (issues de moelle osseuse ou de tissu adipeux) présentaient des anomalies caryotypiques (anomalies de structure) et finissaient par se transformer et exprimer un potentiel oncogénique [23, 24]. Mais les auteurs de ces résultats se sont rétractés : les CSM n’étaient pas responsables du phénotype transformé : en effet, dans tous les cas, les résultats étaient dus à une contamination intralaboratoire par des lignées immortelles cancéreuses épithéliales [25, 26]. De plus, d’autres auteurs ont montré, par une analyse caryotypique ou une analyse par CGH arrays, la stabilité des CSM en culture. Autre argument rassurant, nous avons pu montrer que les CSM cultivées dans des conditions de BPF devenaient sénescentes, arrêtaient de se diviser au cours de la culture et ne se transformaient pas quel que soit le temps de culture et même si une aneuploïdie était constatée au décours de la culture [20].
Un autre risque doit être pris en compte : le risque lié à l’utilisation de CSM sénescentes qui pourraient perdre une partie de leurs fonctions et modifieraient leur profil de sécrétion. Comme cela a été montré pour les fibroblastes par Hayflick dès les années 1960 [27], que ce soit in vitro ou in vivo au sein des tissus, au cours du temps les cellules arrêtent de se diviser et deviennent sénescentes. La sénescence réplicative est due à différents mécanismes : raccourcissement des télomères, activation de la voie de pRB (protéine du rétinoblastome) par le locus INK4a/ARF qui code pour p16ink4a et p19arf, et activation de la voie de p53 [28]. La sénescence et la transformation sont des processus liés ; ainsi les cellules devenant sénescentes peuvent se transformer après une crise transitoire et l’abrogation des mécanismes de sénescence avec une réaugmentation de la longueur des télomères ou la répression de l’activité de p16ink4a et de p53 [22]. Par ailleurs, les cellules devenant sénescentes ont un phénotype sécrétoire spécifique avec production de différentes molécules comme l’IL6 (interleukine 6), des MMP (métalloprotéases), l’hepatocyte growth factor, le FGF2 et GROa (la chimiokine CXCL1). Quoique encore peu exploré, ce phénotype sécrétoire peut agir de façon autocrine et paracrine, renforçant la sénescence, stimulant in vitro et in vivo la croissance de cellules cancéreuses et leur potentiel d’invasion [29, 34], et moduler l’inflammation et la réponse immune. Ainsi, il a été montré dans un modèle murin que les CSM injectées pouvaient faciliter la croissance de la tumeur [30, 36] (→). Il est intéressant de noter dans un modèle de souris présentant des tumeurs mammaires que des CSM dont le gène p53 est muté peuvent migrer spécifiquement au niveau des glandes mammaires et former un microenvironnement permissif pour le développement d’un carcinome mammaire [31]. La sénescence des CSM en culture est un processus continu et compliqué qui est régulé de façon précise au niveau génétique, épigénétique, transcriptionnel et protéique [32]. Il est maintenant prouvé que le risque de transformation des CSM au cours des procédés de culture actuels est faible [33]. Par ailleurs, le caryotype et le CGH arrays, du fait de leur faible sensibilité et du caractère non informatif des anomalies de nombre (aneuploïdie) [20], ne sont pas des contrôles adéquats. Il paraît important, pour juger des risques liés à la transformation (faible) ou à la sénescence, de développer de nouveaux contrôles. Aussi peut-on étudier les transcrits ou les protéines codées par les gènes des voies impliquées dans la sénescence et la transformation comme p53, p21, p16 [20].
(→) Voir l’article de G. Lazennec, page 285 de ce numéro
CSM et utilisation clinique : un monde en constante évolution
Le domaine d’étude et les champs d’application des CSM évoluent rapidement. Depuis le premier essai clinique réalisé en 1995, les CSM cultivées ex vivo ont été utilisées dans de nombreux essais cliniques allant des pathologies immunes (par exemple la réaction du greffon contre l’hôte [GVHD] au cours de l’allogreffe de cellules souches hématopoïétiques) à la médecine régénérative ou à l’ingénierie de l’os, comme le développent C. Jorgensen et al. et C. Vinatier et al. dans ce numéro [37, 38] (→).
(→) Voir pages 275 et 289
Malgré les très nombreux essais cliniques (il y a actuellement 125 essais en cours officiellement répertoriés)5, il n’existe pas de standardisation des procédés de production des CSM. La production de CSM dans des milieux sans produits d’origine animale voire sans sérum et complètement définis est en cours de développement et devra être adaptée à des systèmes clos et automatisés qui répondront parfaitement aux exigences des BPF. De plus, il faut choisir, développer et mettre en place des contrôles standardisés qui permettent d’assurer la sécurité et l’efficacité de l’utilisation des CSM. L’étude des changements complexes de la biologie des CSM au cours de la culture doit permettre de définir des cibles pertinentes pour la sécurité et l’efficacité, en décrire les évolutions potentielles et en faire l’évaluation. Ces études non seulement doivent être faites lors des phases de recherche fondamentale, mais elles doivent également être poursuivies et validées lors des essais cliniques. Ceux-ci doivent se faire selon des règles précises telles qu’éditées par l’International society for stem cell research (ISSCR)6. Il est donc essentiel, pour aborder toutes ces questions touchant à la production de CSM à usage clinique en accord avec les BPF et les contrôles, de développer une interaction forte entre les équipes de recherche, les équipes de R & D, sans oublier les équipes cliniques qui conduisent les essais cliniques (Figure 1).
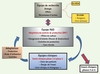 |
Figure 1 Interactions équipes de recherche/équipes de R&D/équipes cliniques. |
Conflit d’intérêts
Les auteurs déclarent n’avoir aucun conflit d’intérêts concernant les données publiées dans cet article.
Remerciements
Ce travail est financé par les programmes CASCADE et REBORNE du 7 e PCRD de la Commission européenne, l’Établissement français du sang, l’Agence de la biomédecine, le Programme hospitalier de recherche clinique (PHRC 2005).
DMEM : Dulbecco’s modified Eagle’s medium ; MEM : minimum essential medium alpha (MEM).
PEI : Paul-Ehrlich-Institut, l’autorité fédérale principale qui relève du ministère fédéral allemand de la Santé.
Voir htpp:/www.isscr.org/clinical trans
Références
- DominiciM, Le BlancK, MuellerI, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The international society for cellular therapy position statement. Cytotherapy 2006 ; 8 : 315-317. [CrossRef] [PubMed] [Google Scholar]
- CrisanM, YapS, CasteillaL, et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 2008 ; 3 : 301-313. [CrossRef] [PubMed] [Google Scholar]
- SensebéL, BourinP. Producing MSC according GMP: process and controls. Biomed Mater Eng 2008 ; 18 : 173-177. [PubMed] [Google Scholar]
- BaxterMA, WynnRF, JowittSN, et al. Study of telomere length reveals rapid aging of human marrow stromal cells following in vitro expansion. Stem Cells 2004 ; 22 : 675-682. [CrossRef] [PubMed] [Google Scholar]
- StolzingA, JonesE, McGonagleD, ScuttA. Age-related changes in human bone marrow-derived mesenchymal stem cells: consequences for cell therapies. Mech Ageing Dev 2008 ; 129 : 163-173. [CrossRef] [PubMed] [Google Scholar]
- KernS, EichlerH, StoeveJ, et al. Comparative analysis of mesenchymal stem cells from bone marrow, umbilical cord blood, or adipose tissue. Stem Cells 2006 ; 24 : 1294-1301. [CrossRef] [PubMed] [Google Scholar]
- BiebackK, KernS, KlüterH, EichlerH. Critical parameters for the isolation of mesenchymal stem cells from umbilical cord blood. Stem Cells 2004 ; 22 : 625-634. [CrossRef] [PubMed] [Google Scholar]
- GronthosS, ZannettinoACW, HaySJ, et al. Molecular and cellular characterisation of highly purified stromal cells derived from human bone marrow. Cell Sci 2003 ; 116 : 1827-1835. [Google Scholar]
- QuiriciN, SoligoD, BossolascoP, et al. Isolation of bone marrow mesenchymal stem cells by anti-nerve growth factor receptor antibodies. Exp Hematol 2002 ; 30 : 783-791. [CrossRef] [PubMed] [Google Scholar]
- DeschaseauxF, CharbordP. Human marrow stromal precursors are alpha 1 integrin subunit-positive. Cell Physiol 2000 ; 184 : 319-325. [CrossRef] [Google Scholar]
- KoçON, GersonSL, CooperBW, et al. Rapid hematopoietic recovery after coinfusion of autologous-blood stem cells and cultured-expanded marrow mesenchymal stem cells in advanced breast cancer patients receiving high-dose chemotherapy. Clin Oncol 2000 ; 18 : 307-316. [Google Scholar]
- ColterDC, SekiyaI, ProckopDJ. Identification of a subpopulation of rapidly self-renewing and multipotential adult stem cells in colonies of human marrow stromal cells. Proc Natl Acad Sci USA 2001 ; 98 : 7841-7845. [CrossRef] [Google Scholar]
- SekiyaI, LarsonBJ, SmithJR, et al. Expansion of human adult stem cells from bone marrow stroma: conditions that maximize the yields of early progenitors and evaluate their quality. Stem Cells 2002 ; 20 : 530-541. [CrossRef] [PubMed] [Google Scholar]
- BanfiA, MuragliaA, DozinB, et al. Proliferation kinetics and differentiation potential of ex vivo expanded human bone marrow stromal cells: Implications for their use in cell therapy. Exp Hematol 2000 ; 28 : 707-715. [CrossRef] [PubMed] [Google Scholar]
- MuragliaA, CanceddaR, QuartoR. Clonal mesenchymal progenitors from human bone marrow differentiate in vitro according to a hierarchical model. Cell Sci 2000 ; 113 : 1161-1166. [Google Scholar]
- NgF, BoucherS, KohS, et al. PDGF, TGF-beta, and FGF signaling is important for differentiation and growth of mesenchymal stem cells (MSCs): transcriptional profiling can identify markers and signaling pathways important in differentiation of MSCs into adipogenic, chondrogenic, and osteogenic lineages. Blood 2008 ; 112 : 295-307. [CrossRef] [PubMed] [Google Scholar]
- MüllerI, KordowichS, HolzwarthC, et al. Animal serum-free culture conditions for isolation and expansion of multipotent mesenchymal stromal cells from human BM. Cytotherapy 2006 ; 8 : 437-444. [CrossRef] [PubMed] [Google Scholar]
- KocaoemerA, KernS, KlüterH, BiebackK. Human AB serum and thrombin-activated platelet-rich plasma are suitable alternatives to fetal calf serum for the expansion of mesenchymal stem cells from adipose tissue. Stem Cells 2007 ; 25 : 1270-1278. [CrossRef] [PubMed] [Google Scholar]
- ChaseLG, LakshmipathyU, SolchagaLA, et al. A novel serum-free medium for the expansion of human mesenchymal stem cells. Stem Cell Res Ther 2010 ; 1 : 8. [CrossRef] [PubMed] [Google Scholar]
- TarteK, GaillardJ, LatailladeJJ, et al. Société française de greffe de moelle et thérapie cellulaire. Clinical-grade production of human mesenchymal stromal cells: occurrence of aneuploidy without transformation. Blood 2010 ; 115 : 1549-1553. [CrossRef] [PubMed] [Google Scholar]
- GodaraP, McFarlandCD, NordonRE. Design of bioreactors for mesenchymal stem cell tissue engineering. Chem Technol Biotechnol 2010 ; 83 : 408-420. [Google Scholar]
- SerakinciN, GuldbergP, BurnsJS, et al. Adult human mesenchymal stem cell as a target for neoplastic transformation. Oncogene 2004 ; 23 : 5095-5098. [CrossRef] [PubMed] [Google Scholar]
- RubioD, Garcia-CastroJ, MartınMC, et al. Spontaneous human adult stem cell transformation. Cancer Res 2005 ; 65 : 3035-3039. [PubMed] [Google Scholar]
- RoslandGV, SvendsenA, TorsvikA, et al. Long-term cultures of bone marrow-derived human mesenchymal stem cells frequently undergo spontaneous malignant transformation. Cancer Res 2009 ; 69 : 5331-5339. [CrossRef] [PubMed] [Google Scholar]
- GarciaS, MartinMC, de la FuenteR, et al. Pitfalls in spontaneous in vitro transformation of human mesenchymal stem cells. Exp Cell Res 2010 ; 316 : 1648-1650. [CrossRef] [PubMed] [Google Scholar]
- TorsvikA, RoslandGV, SvendsenA, et al. Spontaneous malignant transformation of human mesenchymal stem cells reflects cross-contamination: putting the research field on track. Cancer Res 2010 ; 70 : 6393-6396. [CrossRef] [PubMed] [Google Scholar]
- HayflickL. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res 1963 ; 37 : 614-636. [Google Scholar]
- CampisiJ, d’Adda di FagagnaF. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 2007 ; 8 : 729-740. [CrossRef] [PubMed] [Google Scholar]
- CoppéJP, PatilCK, RodierF, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PloS Biol 2008 ; 12 : 2853-2868. [Google Scholar]
- DjouadF, PlenceP, BonyC, et al. Immunosuppressive effect of mesenchymal stem cells favors tumor growth in allogeneic animals. Blood 2003 ; 102 : 3837-3844. [CrossRef] [PubMed] [Google Scholar]
- HoughtonJM, LiH, FanX, et al. Mutations in bone marrow-derived stromal stem cells unmask latent malignancy. Stem Cell Dev 2010 ; 19 : 1153-1166. [CrossRef] [Google Scholar]
- WagnerW, HornP, CastoldiM, et al. Replicative senescence of mesenchymal stem cells: a continuous and organized process. PloS One 2008 ; 3 : 1-12. [PubMed] [Google Scholar]
- ProckopDJ, BrennerM, FibbeWE, et al. Defining the risks of mesenchymal stromal cell therapy. Cytotherapy 2010 ; 12 : 576-578. [CrossRef] [PubMed] [Google Scholar]
- BischofO, DejeanA, PineauP. Une revue de la sénescence cellulaire : ami ou ennemi de la promotion tumorale ?Med Sci (Paris) 2009 ; 25 : 153-160. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- CharbordP, CasteillaL. La biologie des cellules souches mésenchymateuses d’origine humaine. Med Sci (Paris) 2011 ; 27 : 261-268. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- LazennecG. Les cellules souches mésenchymateuses : armes ou dangers pour le traitement des cancers ?Med Sci (Paris) 2011 ; 27 : 285-288. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- JorgensenC, DeschaseauxF, Planat-BenardV, GabisonE. Les cellules souches mésenchymateuses : actualités thérapeutiques. Med Sci (Paris) 2011 ; 27 : 275-284. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- VinatierC, BordenaveL, GuicheuxJ, AmédéeJ. Les cellules souches en ingénierie des tissus ostéoarticulaires et vasculaires. Med Sci (Paris) 2011 ; 27 : 289-296. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1 Interactions équipes de recherche/équipes de R&D/équipes cliniques. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.