")
")
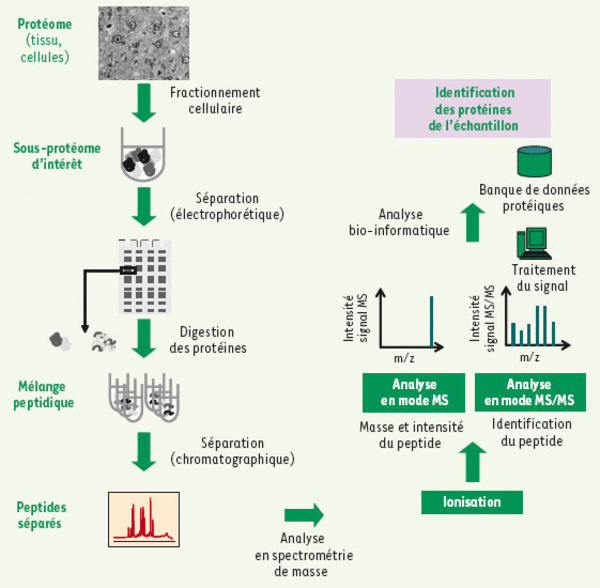
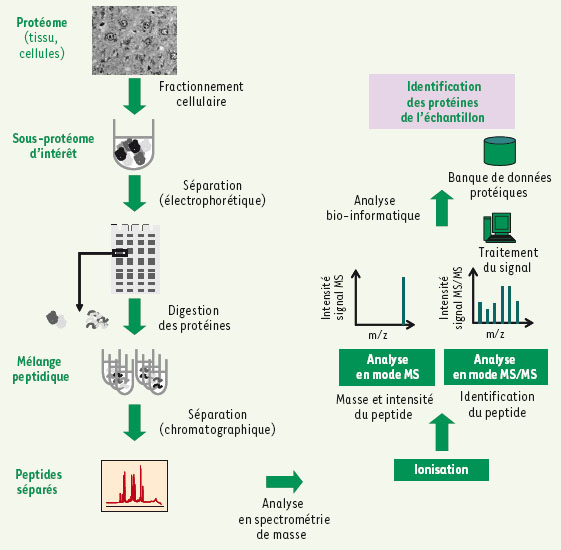
Figure 1.
Télécharger l'image originale
{kind=link}
Déroulement d’une analyse protéomique basée sur la spectrométrie de masse. Une analyse protéomique classique se déroule comme suit : la première étape (à gauche) consiste généralement à fractionner l’échantillon pour le décomplexifier afin de rendre accessible à l’analyse des protéines peu abondantes ou de cibler un complexe protéique. Cette séparation peut être basée sur des méthodes biochimiques (fractionnement subcellulaire, colonnes d’affinité, copurification, etc.) et/ou électrophorétiques (SDS-PAGE ou 2D-GE). Les protéines ainsi séparées sont ensuite digérées par une enzyme protéolytique comme la trypsine. Les peptides générés sont séparés par chromatographie liquide, ionisés, analysés en mode MS afin d’en déduire leur masse (à droite). Les peptides sélectionnés sont fragmentés et analysés en mode MS/MS. Les spectres de fragmentation obtenus sont confrontés avec les spectres MS/MS théoriques obtenus in silico pour chaque peptide trypsique prédit à partir des séquences répertoriées dans une banque de données protéique pertinente. Cette confrontation permet d’identifier la séquence en acides aminés du peptide qui a été fragmenté et, par conséquent, d’identifier la protéine correspondante.
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.