")
")
| Issue |
Med Sci (Paris)
Volume 25, Number 10, Octobre 2009
|
|
|---|---|---|
| Page(s) | 785 - 788 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/20092510785 | |
| Published online | 15 octobre 2009 | |
Mutations du gène TET2 dans les hémopathies myéloïdes humaines
Mutations in TET2 in myeloid cancers
1
E0210 Inserm, Tour Pasteur, Hôpital Necker, 149, rue de Sèvres, 75743 Paris Cedex 15, France
2
Université René Descartes, Faculté de médecine, Paris, France
3
APHP, laboratoire d’hématologie, Hôpital Cochin, F-75014 Paris, France
4
Inserm, UMR790, Institut Gustave Roussy, 39, rue Camille Desmoulins, 94805 Villejuif, France
5
Université Paris XI, F-94805 Villejuif, France
6
Inserm U567, CNRS UMR8104, département d’hématologie, Institut Cochin, Paris, France
7
APHP, laboratoire d’hématologie, Hôpital Saint-Antoine, Paris, France
8
Université Paris VI Pierre et Marie Curie, Paris, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Les hémopathies malignes de type myéloïde sont caractérisées par des modifications des propriétés de survie, de prolifération ou de différenciation des cellules des lignées myéloïdes. Elles sont dues à l’accumulation d’anomalies génétiques généralement acquises qui touchent les cellules souches hématopoïétiques ou des progéniteurs plus ou moins engagés dans une voie de différenciation [1]. Elles sont classées en trois catégories : les leucémies aiguës (LAM), les syndromes myélodysplasiques (SMD) et les syndromes myéloprolifératifs (SMP) (voir encadré). SMD et SMP peuvent évoluer vers une leucémie aiguë [2].
La caractérisation des anomalies de structure chromosomique observées dans les leucémies aiguës humaines a permis l’identification de nombreux gènes qui jouent un rôle majeur dans la différenciation hématopoïétique et qui, modifiés, participent aux processus de transformation [3]. En revanche, bien que la moitié des patients souffrant de SMD présente des anomalies de structure chromosomique, les anomalies génétiques à l’origine ou survenant de façon précoce sont peu connues [4]. La découverte de mutations ponctuelles conférant une activité constitutive à la protéine à activité tyrosine kinase JAK2 a constitué une avancée majeure dans la compréhension du développement des SMP « classiques ». En effet, la mutation JAK2V617F est présente dans pratiquement tous les cas de polyglobulie de Vaquez (PV) et dans la majorité des thrombocytémies essentielles et des myélofibroses primitives [5]. Bien que l’expression de JAK2V617F dans des cellules primaires de souris soit suffisante pour induire une maladie ressemblant à une PV, un faisceau d’arguments suggérait qu’un événement précédant la survenue de JAK2V617F dans l’histoire naturelle de la maladie existait chez certains patients [6].
Le travail publié récemment par nos équipes a permis d’identifier un gène, Ten-Eleven-Translocation (TET)2, muté dans environ 15 % des hémopathies de type myéloïde chez l’homme [7].
Identification des mutations du gène TET2 dans les hémopathies myéloïdes
Deux approches ont été suivies dans cette étude. L’analyse par cytogénétique moléculaire d’une série de six patients porteurs d’une translocation chromosomique touchant le bras long du chromosome 4 (4q24) a permis d’identifier chez tous les patients une région perdue, qui ne contenait que le gène TET2. Chez quatre de ces patients, la perte d’une copie de TET2 a pu être associée à une anomalie de la région codante de TET2 sur la copie restante du gène.
L’analyse par hybridation génomique comparative (CGH) et single nucleotide polymorphism (SNP) array a été réalisée chez cinq patients souffrant de SMP, et présentant au diagnostic une dominance du clone JAK2V617F dans les cellules représentatives des phases précoces de la différenciation hématopoïétique plus marquée que chez la majorité des patients atteints de SMP. Cette étude a mis en évidence une perte d’hétérozygotie du bras long du chromosome 4 chez trois patients. Un de ces trois patients présentait une délétion dans la région 4q24 centrée sur le gène TET2. Une anomalie de la séquence codante de TET2 existait chez deux de ces patients. Le caractère acquis des anomalies de séquence ou de structure de TET2 a pu être montré dans la majorité des cas analysés.
L’analyse de la séquence codante de TET2 dans une série d’échantillons d’hémopathies myéloïdes a montré des variations par rapport aux séquences de référence dans 15/81 (19 %) des SMD, 5/21 (25 %) des LAM secondaires et 24/198 (12 %) des SMP étudiés. Les anomalies de TET2 n’apparaissent pas associées de façon préférentielle avec un sous-groupe de SMD ou de SMP. Les mutations observées étaient majoritairement des mutations tronquantes, par perte ou addition de nucléotides dans la séquence codante du gène ou par mutation non-sens, ou bien des mutations faux-sens touchant des acides aminés conservés dans l’évolution. Dans environ la moitié des échantillons, deux mutations étaient observées, ce qui indique une inactivation complète du gène, mais suggère a contrario que TET2 pourrait avoir un effet à l’état hétérozygote (haplo-insuffisance). Ces données indiquent que TET2 pourrait être un gène de type suppresseur de tumeur muté dans une proportion élevée d’hémopathies humaines de type myéloïde.
Les mutations de TET2 peuvent précéder les mutations de JAK2 dans l’évolution des SMP
La présence des mutations de TET2 dans les progéniteurs hématopoïétiques immatures a ensuite été recherchée. Dans les SMD, les mutations de TET2 sont identifiées dans des cellules primaires triées sur l’expression du marqueur de surface CD34, associé ou non au marqueur CD38, les cellules CD34+CD38- étant considérées comme plus immatures que les cellules CD34+CD38+. Les mutations de TET2 sont détectées dans les cellules progénitrices individuellement amplifiées in vitro. Des observations similaires sont faites avec des échantillons de SMP. L’analyse à l’échelon cellulaire a également permis de montrer dans cinq cas de SMP que les mutations TET2 sont apparues avant la mutation JAK2V617F au cours de l’histoire naturelle de la maladie. En effet des cellules mutées pour TET2 et sauvages pour JAK2 sont observées chez ces patients tandis qu’aucune cellule sauvage pour TET2 et mutée pour JAK2 n’a pu être identifiée.
Pour confirmer que les mutations de TET2 sont présentes dans des cellules ayant des propriétés de cellule souche, des cellules CD34+ isolées chez cinq patients atteints de SMP ont été greffées chez des souris immunodéficientes (NOD-SCID, non-obese diabetic severe combined immunodeficient) : deux échantillons de cellules CD34+ portaient des mutations de TET2, trois avaient un gène TET2 normal. Seules les cellules issues des patients porteurs de la mutation de TET2 étaient capables de proliférer et de se développer chez la souris pendant 15 semaines, entraînant une augmentation progressive du chimérisme (homme/souris). De plus, alors que l’hématopoïèse humaine qui se développe chez les souris greffées avec les échantillons dont le gène TET2 n’est pas muté ou avec des cellules humaines normales est orientée préférentiellement vers une différenciation lymphoïde, l’hématopoïèse humaine observée avec les échantillons porteurs d’un gène TET2 muté est majoritairement de type myéloïde. Si toutes les cellules progénitrices humaines qui sont présentes chez la souris après 15 semaines sont porteuses des mutations TET2, elles ne sont que rarement porteuses de la mutation JAK2V617F. L’ensemble de ces données indique que les mutations de TET2 sont présentes dans des cellules ayant des propriétés de type cellule souche, et leur confèrent un avantage de croissance par rapport aux cellules normales.
Perspectives physiopathologiques pour les hémopathies myéloïdes
Une fréquence élevée de mutations de TET2 a maintenant été rapportée dans diverses pathologies malignes myéloïdes humaines [8–14]. La caractérisation d’un événement oncogénique commun à plusieurs hémopathies myéloïdes pourrait permettre une réévaluation de leur classification. Elle indique, du point de vue physiopathologique, une anomalie commume affectant un stade précoce de l’hématopoïèse, la diversité phénotypique étant ensuite générée par la survenue de différents événements oncogéniques, JAK2V617F pour les SMP ou des mutations de RUNX1 pour les SMD ou les LAM (Figure 1).
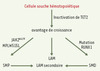 |
Figure 1. Les mutations de TET2 prédisposeraient au développement des hémopathies myéloïdes chez l’homme. Le schéma est hypothétique. Les flèches indiquent des événements oncogéniques qui restent largement à identifier. |
Trois gènes qui codent pour des protéines apparentées à TET2 ont été identifiés chez l’homme. Le membre fondateur TET1 a été isolé lors de la caractérisation d’une protéine de fusion formée avec le produit du gène MLL (myeloid/lymphoid or mixed lineage leukemia) à la suite de la translocation t(10;11)(q22;q23) observée dans des leucémies aiguës [15, 16]. Les protéines de la famille TET sont apparentées aux oxygénases dépendantes des ions ferreux et du 2-oxoglutarate. TET1 est expérimentalement capable d’hydroxyler les 5-méthylcytosines [17]. Cette activité n’était pas connue auparavant chez l’homme et la fonction des 5-hydroxyméthylcytosines reste à déterminer. Néanmoins, ces données suggèrent un rôle dans le contrôle épigénétique de la transcription pour les membres de la famille TET. Ces résultats ouvrent un champ entier de recherche qui concerne les mécanismes de réparation, le contrôle de la transcription, de la différenciation hématopoïétique normale et pathologique et éventuellement la réponse au traitement de ces hémopathies myéloïdes.
Conflit d’intérêts
Les auteurs déclarent n’avoir aucun conflit d’intérêts concernant les données publiées dans cet article.
Remerciements
Le travail présenté ici a été réalisé principalement grâce au soutien de l’INSERM, les universités Paris V et Paris XI, l’INCa, la Ligue nationale contre le cancer et la Fondation de France.
Références
- Huntly BJ, Shigematsu H, Deguchi K, et al. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell 2004; 6 : 587–96. [Google Scholar]
- Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the WHO classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 2009; 8 : 8. [Google Scholar]
- Frohling S, Dohner H. Chromosomal abnormalities in cancer. N Engl J Med 2008; 359 : 722–34. [Google Scholar]
- Nimer SD. Myelodysplastic syndromes. Blood 2008; 111 : 4841–51. [Google Scholar]
- Vainchenker W, Dusa A, Constantinescu SN. JAKs in pathology: role of Janus kinases in hematopoietic malignancies and immunodeficiencies. Semin Cell Dev Biol 2008; 19 : 385–93. [Google Scholar]
- Kralovics R. Genetic complexity of myeloproliferative neoplasms. Leukemia 2008; 22 : 1841–8. [Google Scholar]
- Delhommeau F, Dupont S, Della Valle V, et al. Mutation in TET2 in myeloid cancers. N Engl J Med 2009; 360 : 2289–301. [Google Scholar]
- Tefferi A, Levine RL, Lim KH, et al. Frequent TET2 mutations in systemic mastocytosis: clinical, KITD816V and FIP1L1-PDGFRA correlates. Leukemia 2009; 23 : 900–4. [Google Scholar]
- Tefferi A, Lim KH, Abdel-Wahab O, et al. Detection of mutant TET2 in myeloid malignancies other than myeloproliferative neoplasms: CMML, MDS, MDS/MPN and AML. Leukemia 2009; 23 : 1343–5. [Google Scholar]
- Tefferi A, Pardanani A, Lim KH, et al. TET2 mutations and their clinical correlates in polycythemia vera, essential thrombocythemia and myelofibrosis. Leukemia 2009; 23 : 905–11. [Google Scholar]
- Abdel-Wahab O, Mullally A, Hedvat C, et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood 2009; 114 : 144–7. [Google Scholar]
- Gelsi-Boyer V, Trouplin V, Adelaide J, et al. Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol 2009; 13 : 13. [Google Scholar]
- Jankowska AM, Szpurka H, Tiu RV, et al. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood 2009; 113 : 6403–10. [Google Scholar]
- Langemeijer SM, Kuiper RP, Berends M, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet 2009; 41 : 838–42. [Google Scholar]
- Lorsbach RB, Moore J, Mathew S, et al. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23). Leukemia 2003; 17 : 637–41. [Google Scholar]
- Ono R, Taki T, Taketani T, et al. LCX, leukemia-associated protein with a CXXC domain, is fused to MLL in acute myeloid leukemia with trilineage dysplasia having t(10;11)(q22;q23). Cancer Res 2002; 62 : 4075–80. [Google Scholar]
- Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009; 324 : 930–5. [Google Scholar]
© 2009 médecine/sciences - Inserm / SRMS
Liste des figures
|
Figure 1. Les mutations de TET2 prédisposeraient au développement des hémopathies myéloïdes chez l’homme. Le schéma est hypothétique. Les flèches indiquent des événements oncogéniques qui restent largement à identifier. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.