")
")
| Issue |
Med Sci (Paris)
Volume 24, Number 10, Octobre 2008
|
|
|---|---|---|
| Page(s) | 833 - 840 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/20082410833 | |
| Published online | 15 octobre 2008 | |
Lamines A et syndromes progéroïdes
Une farnésylation persistante aux conséquences dramatiques
A-type Lamins and progeroïd syndromes: persistent farnesylation with dramatic effects
1
Université de la Méditerranée, Inserm UMR 910, « Génétique Médicale et Génomique Fonctionnelle », Faculté de médecine de Marseille 27, boulevard Jean Moulin, 13005 Marseille, France
2
AP-HM, Département de Génétique Médicale, Campus Hospitalo-Universitaire de la Timone, 13385 Marseille Cedex 05, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Les Lamines sont des éléments indispensables de la matrice nucléaire qui participent notamment, au sein de la lamina nucléaire, au maintien de l’intégrité structurale du noyau. La progéria typique de Hutchinson-Gilford (HGPS) est une maladie mimant le vieillissement de façon prématurée et accélérée, entraînant le décès des enfants aux alentours de l’âge de 12 ans. Cette pathologie est causée par une mutation dans le gène LMNA codant les Lamines A/C. Dans la majorité des cas, il s’agit d’une mutation à priori silencieuse, mais qui entraîne en réalité un défaut d’épissage du transcrit primaire aboutissant à la production d’une protéine tronquée. Cette dernière s’accumule sous la forme d’un précurseur farnésylé, ne pouvant plus subir les modifications post-traductionnelles nécessaires à sa maturation. D’autres syndromes progéroïdes ont également été associés à des mutations dans ce même gène comme la Dysplasie Acromandibulaire (MAD) ou plus récemment la Dermopathie Restrictive (RD). Toutes deux peuvent cependant être causées également par des mutations dans le gène FACE1/ZMPSTE24 codant l’une des enzymes majeures responsables de la maturation de la Lamine A. Tous ces syndromes progéroïdes (MAD, HGPS et RD), sont liés à des défauts de maturation de la Lamine A et constituent un continuum phénotypique sous-tendu par un mécanisme physiopathologique commun, en rapport avec l’accumulation d’un précurseur restant anormalement farnésylé. Ce trait commun offre des perspectives de thérapie pharmacologique pour ces pathologies sévères.
Abstract
Hutchinson-Gilford Progeria (HGPS), a rare and severe developmental disorder characterized by features recalling premature aging, and Restrictive Dermopathy (RD), a neonatal lethal genodermatosis, have recently been identified as being primary or secondary « Laminopathies ». These heterogeneous disorders are caused by altered Lamin maturation pathway. In physiological conditions, mature Lamin A is obtained through a series of post-translational processing steps performed on a protein precursor, Prelamin A. The major pathophysiological mechanism involved in Progeria is an aberrant splicing due to a de novo heterozygous point mutation, leading to the accumulation of truncated Lamin A precursor. The same aberrant splicing mechanism was involved in RD, whereas the majority of RD cases are caused by ZMPSTE24/FACE1 inactivation, a key enzyme involved in the Lamin A maturation pathway. In functional terms, all these conditions share the same pathophysiological mechanism, i.e. the intranuclear accumulation of Lamin A precursors, which cannot be fully processed and exert a toxic effect on nuclear homeostasis. In this article, we review the structure and functions of A-type Lamins, focusing namely on HGPS, RD or MAD disorders, in relation to existing animal models and possible future therapeutic approaches.
© 2008 médecine/sciences - Inserm / SRMS
Les Lamines : structure et architecture du noyau
Les Lamines sont les principaux constituants d’un réseau filamenteux situé sous la face nucléoplasmique de l’enveloppe nucléaire et appelé lamina nucléaire. Ces protéines sont en étroite relation à la fois avec l’hétérochromatine ainsi qu’avec de nombreuses protéines associées à l’enveloppe nucléaire. De par leur structure - caractéristique des protéines des filaments intermédiaires - leur permettant de former des hétéropolymères, et leur distribution, majoritairement au niveau de la lamina nucléaire, le seul rôle longtemps attribué aux Lamines a été un rôle de structure. On sait aujourd’hui qu’un pool de Lamines est distribué à l’intérieur du nucléoplasme, que leur dissociation-association est nécessaire lors de la mitose cellulaire, et qu’elles interagissent avec de nombreuses protéines nucléaires, jouant ainsi d’autres rôles dans des phénomènes cruciaux tels que la transcription, l’épissage ou encore la synthèse d’ADN (Figure 1).
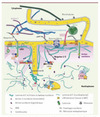 |
Figure 1. Structure du noyau, des Lamines et des principales protéines nucléaires. La lamina nucléaire, constituée de Lamines A/C et B (filaments enchevêtrés verts et épais) est localisée sous la face nucléoplasmique de l’enveloppe nucléaire (EN). Les Lamines A/C sont également présentes dans le nucléoplasme (filaments fins). Elles se lient directement à de nombreuses protéines transmembranaires ancrées dans l’enveloppe nucléaire (LBR, Emerine, LAP2β, MAN1, Nesprines 1 et 2). De plus, elles interagissent avec des protéines qui ne sont pas ancrées à la membrane nucléaire mais y sont étroitement associées (LAP2α), ou encore avec des facteurs de transcription (SREBP1, MOK2), ou d’autres protéines nucléaires, telles que Rb. L’actine est présente sous forme de longs polymères (Actine-F) dans le cytoplasme où elle est reliée aux Nesprines, ancrées dans la face externe de l’EN. Par ailleurs, l’actine est également présente sous forme de monomères (Actine G) ou d’oligomères dans le noyau ou elle est liée aux Lamines et à l’Emerine. Les protéines encadrées (ZYG12 ou GCL) représentent des protéines actuellement non décrites chez l’homme. La Calréticuline, protéine soluble de la lumière de l’EN, permet la visualisation des citernes du réticulum endoplasmique et des quelques invaginations de l’EN à l’intérieur du nucléoplasme. |
Deux types de Lamines existent dans les cellules de mammifères : les Lamines de type A et les Lamines de type B. La distinction la plus importante entre ces deux types est que les Lamines de type B, codées par deux gènes distincts LMNB1 et LMNB2 situés respectivement en 5q.23.3-q.31.1 et en 19p13.3, sont exprimées à tous les stades du développement et sont indispensables à la survie cellulaire, tandis que les Lamines de type A sont codées par le gène LMNA et sont exprimées plus tardivement dans le développement. Le gène LMNA, composé de 12 exons, est localisé en position 1q21.2-q21.3. Il produit quatre transcrits obtenus par épissage alternatif. Les Lamines A et C sont les deux isoformes majeures, tandis que la Lamine A∆10 et la Lamine C2 sont minoritaires et spécifiquement exprimées par certains tissus, dans une lignée tumorale et dans une lignée germinale mâle respectivement. Le site d’épissage alternatif différenciant la Lamine C de la Lamine A se situe dans l’exon 10. La partie du gène codant spécifiquement pour la Lamine A est composée des 90 premières paires de bases de l’exon 10, suivies des exons 11 et 12, tandis que la Lamine C est composée de l’exon 10 dans sa totalité et présente un codon stop à son extrémité 3’ terminale (Figure 2) (pour une revue complète, voir [1]).
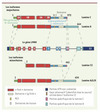 |
Figure 2. Les différentes isoformes protéiques codées par le gène LMNA. Le gène LMNA code pour deux isoformes majoritaires (en haut) et deux isoformes minoritaires (en bas). Les régions 5’ et 3’ UTR non codantes sont représentées en bleu foncé. Le reste du code de couleur est commun à la fois au gène et aux protéines. La partie correspondant au domaine globulaire amino-terminal est indiquée en vert, et la partie carboxy-terminale en bleu clair. La partie spécifique de la Lamine C est également retrouvée dans l’isoforme C2. L’isoforme AΔ10 ne comporte pas les acides aminés codés par l’exon 10 et l’isoforme C2 ne contient pas les acides aminés codés par l’exon 1, mais comprend 6 acides aminés spécifiques, codés par un exon alternatif identifié chez la souris (en marron). |
La Lamine A est d’abord synthétisée sous forme de précurseur, la Pré-lamine A qui, comme les Lamines de type B mais contrairement à la Lamine C, contient un motif de prénylation et plus spécifiquement de farnésylation « CaaX » (C : cystéine ; a : acide aminé aliphatique, X : n’importe quel acide aminé) situé dans sa partie carboxy-terminale [31] (Encadré 1). La présence de ce motif initie une série d’étapes de maturation post-traductionnelle ; on en distingue quatre : (1) l’ajout d’un groupement farnésyl (15 carbones) sur la cystéine de la boîte CaaX par une farnésyl-transférase ; (2) la coupure des trois derniers acides aminés (aaX) par FACE2/RCE1, une endoprotéase ; (3) la fixation d’un groupe méthyl sur la cystéine farnésylée par ICMT, une méthyl-transférase ; (4) le clivage des 15 aminoacides carboxy-terminaux du précurseur par ZMPSTE24/FACE1, une métallo-protéase à zinc, afin d’obtenir la Lamine A mature (Figure 3). La localisation cellulaire de ce processus est encore très discutée dans la communauté scientifique. Nous avons choisi de représenter cette enzyme comme étant ancrée dans la membrane du réticulum endoplasmique (RE) (Figure 3) en nous fondant sur plusieurs études : (1) la localisation dans le RE a été démontrée pour plusieurs enzymes de la voie de maturation des lamines : chez la levure notamment Rce1 et Ste24 sont localisées dans la membrane du RE [2] ; (2) les protéines de la famille Ras sont dirigées vers le RE avant d’être adressées à la membrane plasmique [3] ; (3) ZMPSTE24 comporte, à son extrémité carboxy-terminale, une séquence consensus qui pourrait être un signal de rétention dans le RE. Enfin, (4) il a été démontré spécifiquement pour la lamine B3 que la farnésylation de la lamine B3 s’effectue dans le cytosol, celle-ci étant ensuite importée dans le noyau [4].
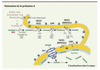 |
Figure 3. Maturation post-traductionnelle de la Prélamine A. Étape 1 : farnésylation de la cystéine carboxy-terminale du motif CAAX (CSIM) par une farnésyl-transférase. Étape 2 : clivage des trois derniers acides aminé aaX, réalisé par une métalloprotéase à zinc, RCE1/FACE2. Étape 3 : carboxyméthylation de la cystéine farnésylée par l’isoprenylcystéine-carboxy-méthyl-transférase (ICMT). Étape 4 : ZMPSTE24/FACE1 réalise le clivage des 15 derniers acides aminés. La Lamine A mature est localisée à la fois sous l’enveloppe nucléaire dans la lamina nucléaire, mais aussi dans le reste du nucléoplasme. |

Contrairement aux Lamines B - le knock-out du gène Lmnb1 est létal au stade embryonnaire - de nombreuses mutations dans le gène LMNA ont été identifiées et sont associées à des phénotypes viables mais de sévérité variable, regroupés sous le terme de « Laminopathies ». À ce jour, plus de 10 Laminopathies différentes ont été identifiées chez l’homme constituant ainsi l’un des modèles les plus caractéristiques d’hétérogénéité clinique des maladies génétiques [1].
Les Laminopathies : un modèle d’hétérogénéité clinique et allélique
La première mutation identifiée dans le gène LMNA responsable d’une forme autosomique dominante de Dystrophie Musculaire d’Emery-Dreifuss (AD-EDMD) a été décrite en 1999 [5]. Brièvement, elle se caractérise par une faiblesse et une atrophie musculaires associées à des rétractions tendineuses et parfois des troubles de la conduction cardiaque. Depuis, une dizaine d’autres pathologies a été associée à des mutations dans LMNA, incluant aussi bien d’autres pathologies musculaires, comme la CMD caractérisée par une atteinte du tissu musculaire cardiaque, que des maladies telles que la FPLD, caractérisée par une fonte du tissu adipeux. Parallèlement, cinq autres gènes codant pour des protéines partenaires des Lamines ont été impliqués dans des pathologies humaines.
Les laminopathies présentent une grande variabilité clinique. Tout d’abord par la diversité des tissus touchés par la pathologie, qui incluent les tissus musculaire, adipeux et osseux. Ceux-ci peuvent l’être de manière isolée, combinée, voire systémique, comme c’est le cas pour la Progéria dans laquelle l’ostéolyse, l’athérosclérose et la lipodystrophie/lipoatrophie sont au premier plan du tableau clinique. Par ailleurs il existe également une hétérogénéité de la sévérité du phénotype, allant d’atteintes modérées chez l’adulte à des formes létales néonatales.
Dans de nombreuses laminopathies (EDMD, FPLD ainsi que dans les syndromes progéroïdes), on observe la présence d’anomalies nucléaires affectant, selon la pathologie, un nombre de noyaux plus ou moins important (allant de 15 % pour EDMD à 50-70 % dans le cas de HGPS ou de la RD). Il s’agit de hernies nucléaires, d’une perte de la condensation de l’hétérochromatine périphérique ou encore de la présence de structures dites en « nid d’abeille », très caractéristiques.
Un certain nombre de mécanismes physiopathologiques ont été suggérés et/ou démontrés afin d’expliquer la grande diversité des pathologies associées à des défauts dans les Lamines, protéines ubiquitaires.
1. En ce qui concerne les laminopathies atteignant spécifiquement les muscles striés squelettique et cardiaque, deux hypothèses sont aujourd’hui avancées et au moins partiellement vérifiées. Selon la première, les mutations associées à ces pathologies déstabilisent la structure tridimensionnelle des Lamines de type A, induisant à terme une perte de la fonction protéique [6]. La seconde suggère que des noyaux dont l’architecture est complètement déformée résistent moins bien au stress mécanique, très important dans ce type tissulaire. En accord avec cette hypothèse, il a été montré que les fibroblastes provenant des souris invalidées pour le gène Lmna ont une faible résistance aux stress mécaniques. Par ailleurs il est possible que d’autres mécanismes soient responsables et/ou associés à celui-ci, comme par exemple un défaut de régénération des fibres musculaires [7].
2. Pour d’autres tissus, tels que le tissu adipeux, il est possible que certains mutants de Lamines puissent perdre ou augmenter leur capacité d’association avec des partenaires moléculaires spécifiques de tissu [8].
3. Pour ce qui est des laminopathies progéroïdes, plusieurs mécanismes pouvant être impliqués simultanément, ont été envisagés [9] : la modification des interactions avec des partenaires plus généraux, tels que certains facteurs de transcription [8, 10] ; une désorganisation de l’hétérochromatine située en périphérie du noyau en étroite relation avec la lamina nucléaire, comme cela a été montré dans des noyaux de patients atteints de HGPS ou MAD ; enfin, un autre mécanisme physiopathologique fondé sur la toxicité de l’accumulation de la Prélamine A farnésylée a été envisagé et sera détaillé ci-après.
Syndromes progéroïdes : la farnésylation, le mécanisme clé ?
La dysplasie acro-mandibulaire (MAD), atteignant particulièrement le tissu osseux, a été l’un des premiers syndromes progéroïdes associé à une mutation homozygote dans le gène LMNA, p.R527H [11]. En se fondant sur des ressemblances phénotypiques entre la progéria classique de Hutchinson-Gilford et le syndrome de MAD, notre équipe a identifié, en 2003, la mutation majeure responsable de la progéria de Hutchinson-Gilford [12]. Dans la majorité des cas de progéria, la mutation en cause est une substitution hétérozygote apparue de novo dans l’exon 11 (c.1864C > T) du gène LMNA. Cette mutation n’entraîne pas de modification prédite de la séquence protéique (p.G608G). En réalité, elle active partiellement un site cryptique d’épissage (encadré 2), délétant les 150 dernières paires de base de l’exon 11 (dont la séquence codant le site de reconnaisance de l’endoprotéase ZMPSTE24/FACE-1), tout en respectant le cadre de lecture de la protéine. La mutation G608G se traduit donc par la production d’une Lamine A tronquée de 50 acides aminés : la Progérine. Celle-ci ne pouvant plus subir la dernière étape de maturation par ZMPSTE24/FACE1, demeure farnésylée sur la dernière cystéine. En 2004, le même type de mécanisme a été mis en évidence dans deux cas de dermopathie restrictive (RD), une génodermatose néonatale très rare caractérisée par un retard de croissance intra-utérin, une peau rigide et épaisse, une arthrogrypose généralisée, une ostéolyse des clavicules et des extrémités ; les enfants décèdent en période néonatale dans un contexte de détresse pulmonaire [13]. La majorité des cas de RD est cependant causée par des mutations dans le gène ZMPSTE24/FACE1 lui-même, à l’état homozygote ou hétérozygote composite, entraînant une absence complète de cette enzyme chez tous les patients testés n’ayant pas de mutation LMNA, et la production exclusive de Prélamine farnésylée [13, 14]. Il existe également une forme de MAD associée à deux mutations hétérozygotes composites dans le gène ZMPSTE24/FACE1, qui cependant n’entraînent qu’une perte partielle de fonction de cette enzyme [15]. Par ailleurs, dans tous les cas de syndromes progéroïdes associés à un défaut de maturation de la Lamine A, on observe, dans une majorité de noyaux, des anomalies nucléaires importantes dont le nombre et la sévérité peuvent être corrélés à la quantité de Prélamine A accumulée [16].

L’identification de la RD comme étant une laminopathie primaire (mutation dans LMNA) ou secondaire (mutations nulles dans ZMPSTE24/FACE1), l’a placée ainsi à l’extrémité la plus sévère du spectre phénotypique de toutes les laminopathies. La caractérisation de la RD a permis d’identifier un mécanisme physiopathologique nouveau et commun à plusieurs syndromes progéroïdes, de sévérité très variable, liés à des défauts de la Lamine A. Dans les cas de mutations de ZMPSTE24/FACE1, on observe une perte de fonction partielle (MAD) ou complète (RD) de la protéine qui a pour conséquence une accumulation plus ou moins importante du précurseur normal, mais farnésylé, dans le noyau. L’observation de cette rétention du groupement farnésyl associée à l’accumulation plus ou moins importante de ce précurseur tronqué (dans le cas de HGPS) ou sauvage (dans les cas de MAD ou RD) soulève l’hypothèse que la toxicité observée dans les noyaux de fibroblastes de tous ces patients peut être liée à la farnésylation persistante de la Prélamine A. Ce mécanisme pourrait donc être à l’origine d’une partie de la spécificité des syndromes progéroïdes, bien qu’il puisse agir de concert avec ceux, soupçonnés ou démontrés comme étant impliqués dans d’autres types de Laminopathies, ce qui expliquerait par ailleurs le chevauchement phénotypique retrouvé dans cet ensemble hétérogène de pathologies.
Apport des modèles animaux et pistes thérapeutiques
L’équipe du Dr Colin Stewart a construit en 1999, dans le but de comprendre la pathophysiologie des laminopathies, une souris knock-out pour le gène Lmna. Le phénotype des souris homozygotes atteint particulièrement les tissus musculaires et graisseux, ainsi que le nerf périphérique, avec une importante réduction de la densité axonale. Il s’agit donc d’un bon modèle pour étudier les laminopathies exprimant une spécificité tissulaire [32]. D’un point de vue cellulaire, l’absence de Lamines A/C cause chez les souris homozygotes des malformations nucléaires, des distributions anormales de plusieurs protéines associées, ainsi que de nombreuses structures chromatiniennes anormales [17]. Les fibroblastes de ces souris montrent une résistance au stress très diminuée par rapport à celle des fibroblastes contrôles, expliquant en partie la pathophysiologie des laminopathies spécifiques du tissu musculaire [7].
Plus récemment, deux modèles d’invalidation génétique de Zmpste24 (Face1), produits de manière indépendante par deux équipes, présentent un phénotype progéroïde très proche de celui de la progéria. Les souris homozygotes ne produisent pas de protéine et miment, au niveau moléculaire et fonctionnel, les anomalies observées chez les patients atteints de RD malgré une létalité post-natale autour de 20 semaines [18, 19]. De nombreuses anomalies nucléaires ont également été identifiées dans les noyaux de fibroblastes de ces souris [18]. Plus récemment, deux études détaillées du phénotype de ces souris ont montré des anomalies de réparation de l’ADN ainsi qu’une instabilité génomique liée à la voie de signalisation p53 [20, 21], reliant ainsi ces nouveaux types de syndromes progéroïdes à des syndromes plus anciennement décrits, tels que le syndrome de Werner, dans lequel le gène impliqué est une hélicase dont la perte fonctionnelle entraîne directement un défaut de réparation de l’ADN [9].
Les croisements réalisés par L.G. Fong, publiés en 2004 et en 2006 [22, 23], ont permis d’identifier un lien entre la quantité de Prélamine accumulée et la sévérité des phénotypes. En effet, les souris résultant du croisement Zmpste24−/− et Lmna+/- ou de croisements Zmpste24−/− et Lmna Lco/+ (des souris ne produisant pas de Lamine A, mais uniquement de la Lamine C) ont montré un phénotype beaucoup moins sévère que celui des souris Zmpste24−/−, Lmna+/+. De récentes approches in vitro, à visée thérapeutique, sont venues confirmer l’hypothèse que nous avions soulevée dans notre étude de cas de RD liés à des mutations dans ZMPSTE24/FACE1. En particulier, la toxicité majeure de la Prélamine A peut provenir de sa farnésylation persistante dans les cas de HGPS ou de RD, et la sévérité des phénotypes dépend étroitement de la quantité de Prélamine A accumulée. Une des premières approches thérapeutiques utilisant des morpholinos dirigés contre le site d’épissage aberrant observé chez les patients atteints de HGPS a montré une diminution des anomalies nucléaires, une amélioration de la condensation de l’hétérochromatine et de plusieurs gènes dérégulés [24]. L’utilisation d’inhibiteurs de farnésyl-transférase (FTI) in vivo semble induire une amélioration du phénotype de ces animaux [25]. Cependant, d’autres pistes ne doivent pas être négligées qui augmenteraient les chances de développer des essais thérapeutiques ciblés sur ces pathologies désastreuses. Notamment parce que les FTI, même s’ils montrent un certain effet bénéfique sur la morphologie nucléaire et les anomalies osseuses des souris traitées, ne modifient pas les anomalies de réparation de l’ADN observées [26]. À ce titre, l’utilisation de molécules inhibitrices de la HMGCoA réductase (statines) et d’inhibiteurs de farnésyl pyrophosphate-synthase de type amino-biphosphonates (N-BPs) semblent de bons candidats. Tout d’abord parce qu’ils se situent en amont de la voie de synthèse du groupement farnésyl ajouté à la Prélamine et qu’ils inhibent à la fois la farnésylation et la géranylgéranylation (qui est l’ajout d’un groupement prényl de type géranyl-géranyl, dérivé du farnésyl). Or cette voie peut être alternativement utilisée lors du blocage de la farnésylation par les FTI ce qui expliquerait leur faible activité (Figure 4) [27]. Par ailleurs, ces deux molécules bénéficient d’une autorisation de mise sur le marché et sont utilisées comme hypolipémiants (pour les statines) chez des patients atteints d’athérosclérose et en prévention ou traitement de l’ostéoporose (pour les N-BPs). Or, ces deux atteintes sont majeures chez les patients HGPS.
 |
Figure 4. Voie de synthèse du cholestérol et inhibiteurs pharmacologiques de prénylation. Les inhibiteurs de prénylation sont représentés en rouge. Les molécules les plus en amont dans la cascade métabolique sont les statines : elles inhibent l’HMGCoA réductase, et diminuent la quantité de substrat pour la farnésyl pyrophosphate-synthase. Celle-ci peut également être inhibée directement par les amino-biphosphonates (N-BP). Enfin, en aval, les inhibiteurs de farnésyl-transférase (FTI) et de géranylgéranyl-transférase (GGTI), inhibent les deux dernières enzymes intervenant dans cette cascade. |
Conclusions
Plusieurs zones d’ombres demeurent quant à la compréhension des mécanismes physiopathologiques responsables des syndromes progéroïdes liés à des défauts de maturation de la Lamine A. Notamment, il ne semble pas que la mutation R527H, causant le syndrome MAD, altère l’épissage de la Lamine, alors que les noyaux cellulaires de ces patients présentent une accumulation de Prélamine A sans que l’on en connaisse exactement le mécanisme. Il en est de même concernant les noyaux de patients atteints de lipodystrophie ou chez certains patients infectés par le VIH et traités avec des inhibiteurs de protéase, qui inhibent aussi ZMPSTE24 [28, 29].
Par ailleurs, des questions de grand intérêt restent ouvertes : comment la farnésylation persistante de la Prélamine A pourrait elle entraîner des dérégulations de mécanismes comme la réparation de l’ADN ? Ou encore comment peut-elle favoriser une instabilité du génome ? Par ailleurs, il a été montré que chez des sujets âgés on observait de la progérine, ce qui permet d’établir un lien, non encore parfaitement caractérisé, entre vieillissement physiologique et prématuré [30]. Il reste beaucoup à apprendre sur ces pathologies grâce à l’exploration complète et minutieuse de patients et de modèles animaux existants ou à venir. Ces derniers permettront également de tester de nouvelles voies thérapeutiques, pharmacologiques, géniques ou cellulaires, en complément ou en alternative aux FTI. Par exemple, l’utilisation isolée ou combinée d’inhibiteurs pharmacologiques de prénylation, agissant en amont dans la voie de maturation des Lamines, pourrait certainement être une voie prometteuse afin d’améliorer les conditions de vie et, peut-être, la survie des enfants atteints de progéria.

Remerciements
Les auteurs tiennent à remercier les docteurs Pierre Cau, Sandrine Pereira, Patrice Bourgeois et Annachiara De Sandre-Giovannoli pour leurs commentaires critiques de ce manuscrit.
Références
- Broers JL, Ramaekers FC, Bonne G, et al. Nuclear lamins : laminopathies and their role in premature ageing. Physiol Rev 2006; 86 : 967–1008. [Google Scholar]
- Schmidt WK, Tam A, Fujimura-Kamada K, et al. Endoplasmic reticulum membrane localization of Rce1p and Ste24p, yeast proteases involved in carboxyl-terminal CAAX protein processing and amino-terminal a-factor cleavage. Proc Natl Acad Sci USA 1998; 95 : 11175–80. [Google Scholar]
- Choy E, Chiu VK, Silletti J, et al. Endomembrane trafficking of ras : the CAAX motif targets proteins to the ER and Golgi. Cell 1999; 98 : 69–80. [Google Scholar]
- Firmbach-Kraft I, Stick R. Analysis of nuclear lamin isoprenylation in Xenopus oocytes : isoprenylation of lamin B3 precedes its uptake into the nucleus. J Cell Biol 1995; 129 : 17–24. [Google Scholar]
- Bonne G, Di Barletta MR, Varnous S, et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet 1999; 21 : 285–8. [Google Scholar]
- Zinn-Justin S. Maladies génétiques et lamines de type A : apport de la biologie structurale. Med Sci (Paris) 2002; 18 : 1054–5. [Google Scholar]
- Lammerding, J. Schulze PC, Takahashi T, et al. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest 2004; 113 : 349–51. [Google Scholar]
- Lloyd DJ, Trembath RC, Shackleton S. A novel interaction between lamin A and SREBP1 : implications for partial lipodystrophy and other laminopathies. Hum Mol Genet 2002; 117 : 769–77. [Google Scholar]
- Navarro CL, Cau P, Levy N. Molecular bases of progeroid syndromes. Hum Mol Genet 2006; 15 : R151–61. [Google Scholar]
- Markiewicz E, Dechat T, Foisner R, et al. Lamin A/C binding protein LAP2alpha is required for nuclear anchorage of retinoblastoma protein. Mol Biol Cell 2002; 1312 : 4401–13. [Google Scholar]
- Novelli, G, Muchir, A, Sangiuolo F, et al. Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C. Am J Hum Genet 2002; 71 : 426–31. [Google Scholar]
- De Sandre-Giovannoli A, Bernard R, Cau P, et al. Lamin a truncation in Hutchinson-Gilford progeria. Science 2003; 300 : 2055. [Google Scholar]
- Navarro CL, Cadinanos J, De Sandre-Giovannoli A, et al. Loss of ZMPSTE24 (FACE-1) causes autosomal recessive restrictive dermopathy and accumulation of Lamin A precursors. Hum Mol Genet 2005; 1411 : 1503–13. [Google Scholar]
- Navarro CL, De Sandre-Giovannoli A, Bernard R, et al. Lamin A and ZMPSTE24 (FACE-1) defects cause nuclear disorganization and identify restrictive dermopathy as a lethal neonatal laminopathy. Hum Mol Genet 2004; 1320 : 2493–503. [Google Scholar]
- Agarwal AK, Fryns JP, Auchus RJ, et al. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum Mol Genet 2003; 1216 : 1995–2001. [Google Scholar]
- Goldman RD, Shumaker DK, Erdos MR, et al. Accumulation of mutant Lamin causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci USA 2004; 101 : 8963–8. [Google Scholar]
- Sullivan T, Escalante-Alcalde D, Bhatt H, et al. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol 1999; 1475 : 913–20. [Google Scholar]
- Bergo MO, Gavino B, Ross J, et al. Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc Natl Acad Sci USA 2002; 99 : 13049–54. [Google Scholar]
- Pendas AM, Zhou Z, Cadinanos J, et al. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat Genet 2002; 311 : 94–9. [Google Scholar]
- Liu B, Wang J, Chan KM, et al. Genomic instability in laminopathy-based premature aging. Nat Med 2005; 117 : 780–5. [Google Scholar]
- Varela I, Cadinanos J, Pendas AM, et al. Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activation. Nature 2005; 437 : 564–8. [Google Scholar]
- Fong LG, Ng JK, Lammerding J, et al. Prelamin A and lamin A appear to be dispensable in the nuclear lamina. J Clin Invest 2006; 116 : 743–52. [Google Scholar]
- Fong LG, Ng JK, Meta M, et al. Heterozygosity for Lmna deficiency eliminates the progeria-like phenotypes in Zmpste24-deficient mice. Proc Natl Acad Sci USA 2004; 101 : 18111–6. [Google Scholar]
- Scaffidi P, Misteli T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat Med 2005; 11 : 440–5. [Google Scholar]
- Fong LG, Frost D, Meta M, et al. A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science 2006; 311 : 1621–3. [Google Scholar]
- Pan J, She M, Xu ZX, et al. Farnesyltransferase inhibitors induce DNA damage via reactive oxygen species in human cancer cells. Cancer Res 2005; 65 : 3671–81. [Google Scholar]
- Van Beek E, Pieterman E, Cohen L, et al. Farnesyl pyrophosphate synthase is the molecular target of nitrogen-containing bisphosphonates. Biochem Biophys Res Commun 1999; 264 : 108–11. [Google Scholar]
- Coffinier C, Hudon SE, Farber EA, et al. HIV protease inhibitors block the zinc metalloproteinase ZMPSTE24 and lead to an accumulation of prelamin A in cells. Proc Natl Acad Sci USA 2007; 104 : 13432–7. [Google Scholar]
- Caron M, Auclair M, Donadille B, et al. Human lipodystrophies linked to mutations in A-type lamins and to HIV protease inhibitor therapy are both associated with prelamin A accumulation, oxidative stress and premature cellular senescence. Cell Death Differ 2007; 14 : 1759–67. [Google Scholar]
- Scaffidi P, Misteli T. Lamin A-dependent nuclear defects in human aging. Science 2006; 312 : 1059–63. [Google Scholar]
- Mazières J, Pradines A, Favre G. Les inhibiteurs de farnésyl transférase : une cible peut en cacher une autre. Med Sci (Paris) 2003; 19 : 211–6. [Google Scholar]
- Langui D, Lachapelle F, Duyckaerts C. Modèles animaux des maladies neuro-dégénératives. Med Sci (Paris) 2007; 23 : 180–6. [Google Scholar]
- Varela I, Pereira S, Ugalde AP, et al. Combined treatment with statins and aminobisphosphonatos extends longevity in a mouse model of human premature aging. Nat Med 2008; 14 : 767–72. [Google Scholar]
- Gilgenkrantz H. Une nouvelle piste thérapeutique dans le traitement des syndromes progéroïdes. Med Sci (Paris) 2008; 24 : 819. [Google Scholar]
Liste des figures
|
Figure 1. Structure du noyau, des Lamines et des principales protéines nucléaires. La lamina nucléaire, constituée de Lamines A/C et B (filaments enchevêtrés verts et épais) est localisée sous la face nucléoplasmique de l’enveloppe nucléaire (EN). Les Lamines A/C sont également présentes dans le nucléoplasme (filaments fins). Elles se lient directement à de nombreuses protéines transmembranaires ancrées dans l’enveloppe nucléaire (LBR, Emerine, LAP2β, MAN1, Nesprines 1 et 2). De plus, elles interagissent avec des protéines qui ne sont pas ancrées à la membrane nucléaire mais y sont étroitement associées (LAP2α), ou encore avec des facteurs de transcription (SREBP1, MOK2), ou d’autres protéines nucléaires, telles que Rb. L’actine est présente sous forme de longs polymères (Actine-F) dans le cytoplasme où elle est reliée aux Nesprines, ancrées dans la face externe de l’EN. Par ailleurs, l’actine est également présente sous forme de monomères (Actine G) ou d’oligomères dans le noyau ou elle est liée aux Lamines et à l’Emerine. Les protéines encadrées (ZYG12 ou GCL) représentent des protéines actuellement non décrites chez l’homme. La Calréticuline, protéine soluble de la lumière de l’EN, permet la visualisation des citernes du réticulum endoplasmique et des quelques invaginations de l’EN à l’intérieur du nucléoplasme. |
| Dans le texte | |
|
Figure 2. Les différentes isoformes protéiques codées par le gène LMNA. Le gène LMNA code pour deux isoformes majoritaires (en haut) et deux isoformes minoritaires (en bas). Les régions 5’ et 3’ UTR non codantes sont représentées en bleu foncé. Le reste du code de couleur est commun à la fois au gène et aux protéines. La partie correspondant au domaine globulaire amino-terminal est indiquée en vert, et la partie carboxy-terminale en bleu clair. La partie spécifique de la Lamine C est également retrouvée dans l’isoforme C2. L’isoforme AΔ10 ne comporte pas les acides aminés codés par l’exon 10 et l’isoforme C2 ne contient pas les acides aminés codés par l’exon 1, mais comprend 6 acides aminés spécifiques, codés par un exon alternatif identifié chez la souris (en marron). |
| Dans le texte | |
|
Figure 3. Maturation post-traductionnelle de la Prélamine A. Étape 1 : farnésylation de la cystéine carboxy-terminale du motif CAAX (CSIM) par une farnésyl-transférase. Étape 2 : clivage des trois derniers acides aminé aaX, réalisé par une métalloprotéase à zinc, RCE1/FACE2. Étape 3 : carboxyméthylation de la cystéine farnésylée par l’isoprenylcystéine-carboxy-méthyl-transférase (ICMT). Étape 4 : ZMPSTE24/FACE1 réalise le clivage des 15 derniers acides aminés. La Lamine A mature est localisée à la fois sous l’enveloppe nucléaire dans la lamina nucléaire, mais aussi dans le reste du nucléoplasme. |
| Dans le texte | |
|
Figure 4. Voie de synthèse du cholestérol et inhibiteurs pharmacologiques de prénylation. Les inhibiteurs de prénylation sont représentés en rouge. Les molécules les plus en amont dans la cascade métabolique sont les statines : elles inhibent l’HMGCoA réductase, et diminuent la quantité de substrat pour la farnésyl pyrophosphate-synthase. Celle-ci peut également être inhibée directement par les amino-biphosphonates (N-BP). Enfin, en aval, les inhibiteurs de farnésyl-transférase (FTI) et de géranylgéranyl-transférase (GGTI), inhibent les deux dernières enzymes intervenant dans cette cascade. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.