")
")
| Issue |
Med Sci (Paris)
Volume 24, Number 6-7, Juin-Juillet 2008
|
|
|---|---|---|
| Page(s) | 659 - 664 | |
| Section | Recherche et partenariat | |
| DOI | https://doi.org/10.1051/medsci/20082467659 | |
| Published online | 15 juin 2008 | |
Biodisponibilité et vecteurs particulaires pour la voie orale
Oral bioavailability and drug/carrier particulate systems
1
Technologie Servier, 25/27, rue Eugène Vignat, 45000 Orléans, France
2
Inserm ERIT-M 0104, Ingénierie de la Vectorisation Particulaire, Immeuble IBT, Université d’Angers, 10, rue André Boquel, 49100 Angers, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Certaines nouvelles molécules à fort potentiel thérapeutique ne sont pas développées en raison de leurs propriétés physico-chimiques qui limitent la fraction circulant sous forme active après administration orale. Une alternative technologique consiste à piéger ces molécules dans des vecteurs particulaires afin de modifier leur devenir in vivo et d’améliorer leurs performances. Des résultats précliniques prometteurs ont été obtenus grâce à cette technologie qui rend envisageable l’administration orale de ces principes actifs. L’enjeu consiste maintenant à optimiser ces assemblages principe actif/transporteur afin d’améliorer encore les propriétés biopharmaceutiques de ces molécules tout en concevant des procédés de fabrication simples et industrialisables.
Abstract
The oral route remains the preferred route of administration to ensure patient satisfaction and compliance. However, new chemical entities may exhibit low bioavailability after oral administration because of poor stability within the gastrointestinal tract, poor solubility in gastrointestinal fluids, low mucosal permeability, and/or extensive first-pass metabolism. Consequently, these new drug substances cannot be further developed using conventional oral formulations. This issue is addressed by an innovative approach based on the entrapment of drug molecules in drug/carrier assembling systems. The carrier materials are lipids, naturally occurring polymers or synthetic polymers, which are considered as nontoxic and biocompatible materials. Drug entrapment is intended to protect drug substances against degradation by gastrointestinal fluids. Fine drug/carrier particle size ensures increased drug dissolution rates. Carriers and particle supramolecular organization can be designed to enhance drug absorption through the intestinal epithelium and lymphatic transport. Promising preclinical results have been obtained with model drugs like paclitaxel, insulin, calcitonin, or cyclosporin. Attention has focused on mucoadhesive carriers like chitosan that favor an intimate and extended contact between drugs and intestinal cells, thus enhancing absorption. Addition of ligands such as lectins improves intestinal drug absorption through specific binding of the carrier to intestinal cell carbohydrates. In conclusion, drug/carrier particulate systems are an attractive and exciting drug delivery strategy for highly potent drug substances unsuitable for oral use. Further evidence will determine whether this approach has marked therapeutic benefits over conventional drug formulations and is compatible with large-scale industrial production and stringent registration requirements. Producing highly effective particulate systems requiring low-complexity manufacturing processes is therefore an ongoing challenge.
© 2008 médecine/sciences - Inserm / SRMS
La voie orale est la voie d’administration de choix des médicaments car elle est la plus naturelle et la plus confortable pour le patient, permettant de garantir au mieux le suivi des prescriptions. Toutefois, l’administration orale d’un nombre important de nouvelles molécules actives issues des étapes de screening pharmacologique révèle une faible biodisponibilité d’après les premières études pharmacocinétiques. Le potentiel thérapeutique de ces molécules est en fait limité par leurs caractéristiques physicochimiques, peu favorables à leur passage dans la circulation systémique. Certaines sont peu stables en milieu physiologique et sont rapidement dégradées dans l’environnement acide de l’estomac ainsi que sous l’action des nombreuses enzymes présentes tout au long du tractus digestif (peptides et protéines thérapeutiques). D’autres sont très peu solubles en milieu aqueux et/ou peu perméables vis-à-vis des membranes biologiques et sont donc mal absorbées au niveau de la muqueuse intestinale. Enfin, la mise en évidence d’un important effet de premier passage hépatique peut stopper le développement d’une nouvelle molécule dès les phases précliniques.
Diverses solutions sont proposées afin d’améliorer la biodisponibilité orale des médicaments. Ainsi, la dégradation dans les fluides digestifs peut être limitée par l’administration simultanée d’inhibiteurs qui diminuent l’activité enzymatique du tractus gastro-intestinal. La vitesse de dissolution des molécules peu solubles peut être améliorée en réduisant la taille des particules de principe actif et en augmentant ainsi leur surface spécifique, grâce à des stratégies telles que la micronisation, le broyage en milieu humide ou la précipitation contrôlée. Le passage à travers la muqueuse intestinale peut également être favorisé par l’utilisation de promoteurs d’absorption qui augmentent de façon transitoire la perméabilité de l’épithélium. Enfin, dans le cas de principes actifs caractérisés par une fenêtre d’absorption, une approche reposant sur des systèmes gastro-résidants ou des systèmes à libération programmée peut favoriser une délivrance site-spécifique dans le tractus gastro-intestinal. Ces différentes stratégies permettent ainsi de dépasser individuellement chacune des limites liées aux caractéristiques physico-chimiques du principe actif et peuvent être éventuellement combinées. Cependant, elles ne sont pas sans inconvénients, surtout dans le cadre de thérapies chroniques. Ainsi, l’administration d’inhibiteurs enzymatiques affecte tout le processus de la digestion et peut à la fois engendrer une mauvaise assimilation des nutriments et empêcher la dégradation d’agents pathogènes. L’action des promoteurs d’absorption n’est quant à elle pas spécifique et favorise le transport dans la circulation sanguine non seulement du principe actif mais également des molécules potentiellement toxiques présentes dans la lumière intestinale.
Une alternative prometteuse consiste à piéger le principe actif au sein d’un vecteur particulaire dont la taille est comprise entre quelques dizaines de nanomètres et quelques centaines de microns. Ces particules sont dispersées dans un véhicule liquide approprié et administrées par voie orale. Le vecteur remplit des rôles multiples : (1) il assure la protection de la molécule active vis-à-vis des dégradations dans le tractus gastro-intestinal en l’isolant des milieux biologiques traversés ; (2) il augmente la cinétique de dissolution du principe actif en le présentant sous une forme finement dispersée ; (3) il peut favoriser l’absorption du principe actif à travers la muqueuse intestinale. Ainsi, la recherche galénique s’intéresse tout particulièrement à la construction de nouveaux systèmes principe actif/transporteur, assemblés sous forme de particules le plus souvent submicroniques et susceptibles de développer des interactions spécifiques avec les membranes biologiques afin d’améliorer la performance des médicaments.
Les vecteurs particulaires à base de lipides
Les vecteurs doivent respecter certaines contraintes pour un usage pharmaceutique. Ils doivent être composés de matériaux biocompatibles n’entraînant aucune réaction toxique, immunogène ou inflammatoire, et doivent être rapidement éliminés par l’organisme afin d’éviter toute accumulation qui pourrait être toxique à long terme. Les formulations à base de lipides (liposomes, nanoparticules et nanocapsules lipidiques, Figure 1) présentent donc un atout majeur puisqu’elles sont constituées essentiellement de composés peu ou pas toxiques.
 |
Figure 1. Vecteurs particulaires utilisés pour la délivrance de médicaments. Liposomes(A), nanocapsules lipidiques(B), nanosphères de polymère (C), nanocapsules de polymère(D). |
Les liposomes sont facilement déstabilisés dans le tractus digestif et sont donc jugés peu performants par voie orale. Certains « liposomologistes » proposent néanmoins de stabiliser ces particules en polymérisant la bicouche phospholipidique qui constitue leur paroi, en les recouvrant d’un polymère hydrophile, ou encore en sélectionnant une composition spécifique de lipides. L’administration orale de liposomes recouverts de chitosane et contenant de la calcitonine à des rats permet ainsi de diminuer leur calcémie de 15% alors qu’aucun effet thérapeutique n’est détecté lorsque la calcitonine est administrée seule en solution [1].
Les nanoparticules solides à base de lipides [2] et les nanocapsules de lipides [3] montrent également un effet sur la biodisponibilité orale de certains principes actifs. Peltier et al. [4] décrivent que l’administration orale de nanocapsules de lipides chargées en paclitaxel multiplie par trois la concentration plasmatique du principe actif en comparaison d’une administration de paclitaxel sous forme non encapsulée.
Au sens strict, les émulsions et les systèmes auto-émulsifiants ne tombent pas dans la catégorie des vecteurs particulaires. En effet, le principe actif est contenu dans une gouttelette liquide de corps gras et non dans une particule solide ou semi-solide. Toutefois, les succès commerciaux de ces formulations témoignent de l’intérêt d’une présentation sous forme dispersée pour des principes très lipophiles : Neoral® (cyclosporine A), Norvir® (ritonavir) ou Fortovase® (saquinavir). La formulation auto-émulsifiante Neoral® multiplie par 2 la biodisponibilité de la cyclosporine par rapport à sa formulation antérieure Sandimmun®, la biodisponibilité absolue atteignant alors 40 % [5]. La formulation du saquinavir Fortovase®, un inhibiteur de protéase du VIH, permet également d’augmenter la biodisponibilité d’un facteur 4 par rapport à une formulation classique. Cet effet est attribué à l’incorporation dans la formulation d’un glycéride qui permet de solubiliser de grandes quantités de principe actif puis de le libérer rapidement in vivo.
L’efficacité revendiquée des formulations à base de lipides repose sur leur capacité à promouvoir l’absorption mucosale des molécules actives. Cependant, la présentation du principe actif sous forme finement dispersée et la stimulation du transport lymphatique ont probablement également un rôle déterminant.
Les vecteurs particulaires à base de polymères
De nombreuses formulations à base de polymères ont également été proposées. La structure des particules polymères chargées en principe actif dépend du procédé de fabrication utilisé, de la nature des matériaux polymères et du caractère lipo- ou hydrosoluble du principe actif. Les nanosphères, constituées d’une matrice polymère continue dans laquelle le principe actif est uniformément dispersé, se distinguent des nanocapsules, qui correspondent à des systèmes réservoirs où un cœur aqueux (encapsulation de molécules hydrophiles) ou huileux (encapsulation de molécules lipophiles) est entouré d’une fine membrane polymère (Figure 1). Les polymères employés pour la conception de vecteurs pharmaceutiques peuvent être issus de la synthèse chimique ou être d’origine naturelle.
Les polymères synthétiques les plus largement utilisés sont les poly(cyanoacrylates d’alkyles) [6], les copolymères des acides lactique et glycolique [7] et la poly-e-caprolactone [8] (Tableau I). Damgé et al. [9] ont démontré que l’administration intragastrique de nanocapsules de poly(cyanoacrylate d’isobutyle) contenant de l’insuline permet de réduire la glycémie de rats diabétiques de 50 à 60 %, alors qu’aucun effet thérapeutique n’est observé lorsque l’insuline est administrée en solution. L’administration orale de nanoparticules de poly-e-caprolactone contenant de la cyclosporine permet également de doubler la dose de principe actif présente dans la circulation systémique ainsi que dans divers organes tels que le foie, les reins ou la rate [10].
Formules chimiques des principaux polymères utilisés pour la conception de vecteurs pharmaceutiques.
Les polymères d’origine naturelle retenus sont généralement des polysaccharides ayant des propriétés gélifiantes comme le chitosane [11], l’alginate [12] ou l’acide hyaluronique [13] (Tableau I). La fabrication des particules fait appel à des techniques douces et des conditions opératoires non dénaturantes pour les principes actifs (milieu aqueux, température ambiante, agitation modérée). L’alginate a ainsi la capacité de former en milieu aqueux des particules gélifiées en présence de cations divalents comme le calcium. Ces gels sensibles au pH ont été très étudiés pour la délivrance de principes actifs par voie orale du fait de leur comportement en milieu gastro-intestinal : ils se rétractent à pH acide, protégeant les principes actifs fragiles des dégradations dans l’estomac, puis gonflent à pH neutre, libérant ainsi leur cargaison en milieu intestinal. Sarmento et al. [14] ont montré que l’administration de nanoparticules d’alginate recouvertes de chitosane et contenant de l’insuline permet d’augmenter la biodisponibilité du principe actif d’un facteur 4. Les particules sub-microniques de chitosane sont quant à elles obtenues par gélification ionique [15] ou par coacervation complexe avec des polyanions comme le sulfate de dextrane [16] ou l’acide hyaluronique [17]. L’administration orale de particules de chitosane contenant de l’insuline à des rats diabétiques permet ainsi d’obtenir un effet hypoglycémiant prolongé et une biodisponibilité relative de l’insuline de 14,9 % par rapport à une injection sous-cutanée [15].
Les vecteurs particulaires adhésifs
L’utilisation de vecteurs bio- ou muco-adhésifs permet d’immobiliser le principe actif à la surface de la muqueuse intestinale et d’augmenter ainsi la fraction de principe actif absorbé. La libération du principe actif se fait alors au plus proche de son site d’absorption, créant un gradient de concentration local qui favorise son passage à travers l’épithélium intestinal. Cette proximité permet également de limiter la dégradation du principe actif dans le fluide luminal.
L’immobilisation du vecteur et de sa cargaison au niveau de la muqueuse intestinale peut se faire soit en surface des cellules épithéliales (bioadhésion) soit au sein du mucus qui recouvre la quasi-intégralité de l’épithélium intestinal (mucoadhésion). Le mucus est un gel visco-élastique formé de glycoprotéines hydrophiles de haute masse molaire et chargées négativement, appelées mucines. L’adsorption d’un vecteur particulaire à la surface de l’intestin résulte des interactions qui s’établissent entre les groupements fonctionnels présents en surface du vecteur et ces glycoprotéines. Ces interactions sont de différentes natures : interactions électrostatiques, interactions hydrophobes, interactions de Van der Waals ou liaisons hydrogène. Le caractère muco-adhésif d’un vecteur dépend donc de ses propriétés de surface, en particulier de sa charge et de son hydrophobie [18]. Les polymères hydrophiles flexibles et de haute masse molaire comme le chitosane et ses dérivés [19], le poly(éthylène glycol) [20], ou encore le poly(acide acrylique) [21] interagissent fortement avec le mucus [22]. Bernkop-Schnürch et al. [23] ont montré que les dérivés thiolés de ces polymères ont la capacité de former des liaisons covalentes (ponts disulfure) avec les régions riches en cystéine du mucus. Ainsi, le couplage d’une cystéine sur du chitosane, du poly(acide acrylique) ou de l’alginate augmente leurs propriétés muco-adhésives de 2 à 140 fois.
Certains auteurs proposent de greffer des ligands en surface des particules afin de cibler spécifiquement certains récepteurs membranaires exprimés au niveau des cellules épithéliales de l’intestin. Les ligands les plus utilisés sont les lectines [24], la vitamine B12 [25] et les invasines [26]. Le greffage de lectines sur des nanoparticules de poly(acide lactique-co-acide glycolique) contenant de la thymopentine, un pentapeptide synthétique, permet ainsi d’améliorer son absorption intestinale de plus d’un facteur 2 par rapport à l’administration de nanoparticules non conjuguées [27].
Enfin, le contact étroit entre le vecteur et les cellules épithéliales pourrait mener à sa propre internalisation dans la muqueuse intestinale. L’utilisation d’un vecteur particulaire augmenterait ainsi l’absorption des molécules actives non seulement en les immobilisant et en les concentrant sur leur site d’absorption préférentiel, mais également en les transportant à travers les membranes biologiques. Cependant, aucun consensus clair n’est établi concernant le devenir in vivo des vecteurs particulaires et leur capacité à traverser les membranes biologiques tout en conservant leur intégrité et en retenant leur cargaison.
L’absorption intestinale des vecteurs particulaires
Le passage des vecteurs particulaires à travers l’épithélium intestinal peut se faire par trois voies différentes : la diffusion entre deux cellules épithéliales adjacentes (voie paracellulaire) [28], l’absorption par les entérocytes (voie transcellulaire) [29] ou la capture par les cellules M présentes dans les tissus lymphoïdes associés à l’intestin (voie lymphatique) [30] (Figure 2). Suite à ce passage de l’épithélium, de nombreuses études montrent que la voie d’absorption prédominante des systèmes particulaires est la voie lymphatique [31] mais que la fraction de particules absorbées sous forme intacte demeure, quoiqu’il en soit, marginale [32].
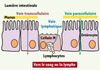 |
Figure 2. Le passage des systèmes particulaires à travers l’épithélium intestinal. L’épithélium intestinal est formé d’une monocouche de cellules étroitement liées les unes aux autres par des jonctions serrées. Le principe actif ou le vecteur pharmaceutique peuvent emprunter trois voies de passage à travers cet épithélium afin de quitter la lumière intestinale et rejoindre la circulation sanguine ou lymphatique : la voie paracellulaire (passage entre deux cellules adjacentes), la voie transcellulaire (absorption par les entérocytes, cellules spécialisées dans l’absorption des nutriments) et la voie lymphatique (absorption par les cellules M associées aux follicules lymphoïdes de l’intestin). |
L’internalisation des nanoparticules dans la muqueuse intestinale peut être favorisée par le choix de certaines de leurs caractéristiques comme leur taille, leur charge de surface ou leur hydrophobie, qui influent sur la nature et la force des interactions entre le vecteur et l’épithélium [18]. Les études menées jusqu’à présent n’ont pas permis d’établir les caractéristiques du vecteur idéal, certains résultats quelque peu contradictoires ayant été décrits dans la littérature. Cependant, les auteurs s’accordent à dire que les vecteurs de petite taille (de préférence inférieure à 200 nm de diamètre) sont absorbés en plus grande proportion.
Récemment la société Sporomex a proposé un nouveau système particulaire reposant sur des spores d’origine végétale capables de piéger des quantités importantes de principe actif par des procédés d’encapsulation simples. Des études en microscopie de fluorescence ont permis de signaler la présence de spores intactes dans la circulation sanguine après administration chez l’homme. Des essais complémentaires doivent cependant être menés afin de confirmer la capacité de ces particules d’une vingtaine de microns à traverser de manière significative la muqueuse digestive.
Conclusion et perspectives
L’administration orale des principes actifs peu solubles, peu perméables à travers les membranes biologiques ou peu stables dans les milieux physiologiques, est l’un des grands défis technologiques qui animent les laboratoires de formulation académiques et industriels. Les différentes études menées jusqu’à présent montrent que l’utilisation de vecteurs particulaires permet d’augmenter de manière significative la biodisponibilité de ces principes actifs. Le mécanisme est sans doute multifactoriel et incomplètement élucidé. Au regard des nombreux articles de recherche scientifiques publiés, des études de preuve de concept chez l’animal et de plusieurs études chez l’homme, la mise sur le marché de nouveaux médicaments pour la voie orale reposant sur une stratégie de vectorisation particulaire devrait devenir une réalité dans les prochaines années. Mais ces technologies innovantes doivent sans doute démontrer une amélioration nette du service médical rendu pour des principes actifs à fort potentiel, tout en conservant un niveau de complexité compatible avec l’industrialisation du procédé de fabrication et les exigences d’une procédure d’enregistrement auprès des autorités réglementaires.

Références
- Takeuchi H, Matsui Y, Sugihara H, et al. Effectiveness of submicron-sized, chitosan-coated liposomes in oral administration of peptide drugs. Int J Pharm 2005; 303 : 160–70. [Google Scholar]
- Zur Muhlen A, Schwarz C, Mehnert W. Solid lipid nanoparticles (SLN) for controlled drug delivery - Drug release and release mechanism. Eur J Pharm Biopharm 1998; 45 : 149–55. [Google Scholar]
- Lamprecht A, Saumet JL, Roux J, Benoit JP. Lipid nanocarriers as drug delivery system for ibuprofen in pain treatment. Int J Pharm 2004; 278 : 407–14. [Google Scholar]
- Peltier S, Oger JM, Lagarce F, et al. Enhanced oral paclitaxel bioavailability after administration of paclitaxel-loaded lipid nanocapsules. Pharm Res 2006; 23 : 1243–50. [Google Scholar]
- Gao ZG, Choi HG, Shin HJ, et al. Physicochemical characterization and evaluation of a microemulsion system for oral delivery of cyclosporin A. Int J Pharm 1998; 161 : 75–86. [Google Scholar]
- Couvreur P. Polyalkylcyanoacrylates as colloidal drug carriers. Crit Rev Ther Drug Carrier Syst 1988; 5 : 1–20. [Google Scholar]
- Italia JL, Bhatt DK, Bhardwaj V, et al. PLGA nanoparticles for oral delivery of cyclosporine: Nephrotoxicity and pharmacokinetic studies in comparison to Sandimmune Neoral. J Control Release 2007; 119 : 197–206. [Google Scholar]
- Hombreiro Perez M, Zinutti C, Lamprecht A, et al. The preparation and evaluation of poly([epsilon]-caprolactone) microparticles containing both a lipophilic and a hydrophilic drug. J Control Release 2000; 65 : 429–38. [Google Scholar]
- Damgé C, Michel C, Aprahamian M, Couvreur P. New approach for oral administration of insulin with polyalkylcyanoacrylate nanocapsules as drug carrier. Diabetes 1988; 37 : 246–51. [Google Scholar]
- Varela MC, Guzman M, Molpeceres J, et al. Cyclosporine-loaded polycaprolactone nanoparticles : immunosuppression and nephrotoxicity in rats. Eur J Pharm Sci 2001; 12 : 471–8. [Google Scholar]
- Alonso MJ, Prego C, Garcia-Fuentes M. Polysaccharide-based nanoparticles as carriers for drug and vaccine delivery. In : Domb AJ, Tabata Y, Kumar MNV, Farber S, eds. Nanoparticles for pharmaceutical applications. New York : American Scientific Publishers, 2007 : 135–50. [Google Scholar]
- Rajaonarivony M, Vauthier C, Couarraze G, et al. Development of a new drug carrier made from alginate. J Pharm Sci 1993; 82 : 912–8. [Google Scholar]
- Jedestrom G, Andersson A, Grasjö J, Sjöholm I. Formulating insulin for oral administration : preparation of hyaluronan-insulin complex. Pharm Res 2004; 21 : 2040–7. [Google Scholar]
- Sarmento B, Ribeiro A, Veiga F, et al. Alginate/chitosan nanoparticles are effective for oral insulin delivery. Pharm Res 2007; 24 : 2198–206. [Google Scholar]
- Pan Y, Li YJ, Zhao HY, et al. Bioadhesive polysaccharide in protein delivery system: chitosan nanoparticles improve the intestinal absorption of insulin in vivo. Int J Pharm 2002; 249 : 139–47. [Google Scholar]
- Janes KA, Fresneau MP, Marazuela A, et al. Chitosan nanoparticles as delivery systems for doxorubicin. J Control Release 2001; 73 : 255–67. [Google Scholar]
- Wawrezinieck A. Élaboration d’un nouveau vecteur pharmaceutique complexe pour l’administration par voie orale d’un peptide protecteur contre le lupus érythémateux disséminé. Thèse pour l’obtention du grade de docteur de l’université Louis Pasteur de Strasbourg, 2007. [Google Scholar]
- Jung T, Kamm W, Breitenbach A, et al. Biodegradable nanoparticles for oral delivery of peptides : is there a role for polymers to affect mucosal uptake ? Eur J Pharm Biopharm 2000; 50 : 147–60. [Google Scholar]
- Bravo-Osuna I, Vauthier C, Farabollini A, et al. Mucoadhesion mechanism of chitosan and thiolated chitosan-poly(isobutyl cyanoacrylate) core-shell nanoparticles. Biomaterials 2007; 28 : 2233–43. [Google Scholar]
- Bertholon I, Ponchel G, Labarre D, et al. Bioadhesive properties of poly(alkylcyanoacrylate) nanoparticles coated with polysaccharide. J Nanosci Nanotechnol 2006; 6 : 3102–9. [Google Scholar]
- Park K, Robinson JR. Bioadhesive polymers as platforms for oral-controlled drug delivery : method to study bioadhesion. Int J Pharm 1984; 19 : 107–27. [Google Scholar]
- Ponchel G, Montisci MJ, Dembri A, et al. Mucoadhesion of colloidal particulate systems in the gastro-intestinal tract. Eur J Pharm Biopharm 1997; 44 : 25–31. [Google Scholar]
- Bernkop-Schnürch A. Thiomers : a new generation of mucoadhesive polymers. Adv Drug Deliv Rev 2005; 57 : 1569–82. [Google Scholar]
- Montisci MJ, Dembri A, Giovannuci G, et al. Gastrointestinal transit and mucoadhesion of colloidal suspensions of lycopersicon esculentum L. and lotus tetragonolobus lectin-PLA microsphere conjugates in rats. Pharm Res 2001; 18 : 829–37. [Google Scholar]
- Russell-Jones GJ, Arthur L, Walker H. Vitamin B12-mediated transport of nanoparticles across Caco-2 cells. Int J Pharm 1999; 179 : 247–55. [Google Scholar]
- Hussain N, Florence AT. O-3 Invasin-induced oral uptake of nanospheres: utilising bacterial mechanisms of epithelial cell entry. J Control Release 1996; 41 : S3–4. [Google Scholar]
- Yin Y, Chen D, Qiao M, et al. Lectin-conjugated PLGA nanoparticles loaded with thymopentin: Ex vivo bioadhesion and in vivo biodistribution. J Control Release 2007; 123 : 27–38. [Google Scholar]
- Aprahamian M, Michel C, Humbert W, et al. Transmucosal passage of polyalkylcyanoacrylate nanocapsules as a new drug carrier in the small intestine. Biol Cell 1987; 61 : 69–76. [Google Scholar]
- Jani PU, Florence AT, McCarthy DE. Further histological evidence of the gastrointestinal absorption of polystyrene nanospheres in the rat. Int J Pharm 1992; 84 : 245–52. [Google Scholar]
- Eldridge JH, Hammond CJ, Meulbroek JA, et al. Controlled vaccine release in the gut-associated lymphoid tissues. I. Orally administered biodegradable microspheres target the peyer’s patches. J Control Release 1990; 11: 205–14. [Google Scholar]
- Jenkins PG, Howard KA, Blackball NW, et al. Microparticulate absorption from the rat intestine. J Control Release 1994; 29 : 339–50. [Google Scholar]
- Ponchel G, Irache JM. Specific and non-specific bioadhesive particulate systems for oral delivery to the gastro-intestinal tract. Adv Drug Deliv Rev 1998; 44 : 25–31. [Google Scholar]
Liste des tableaux
Formules chimiques des principaux polymères utilisés pour la conception de vecteurs pharmaceutiques.
Liste des figures
|
Figure 1. Vecteurs particulaires utilisés pour la délivrance de médicaments. Liposomes(A), nanocapsules lipidiques(B), nanosphères de polymère (C), nanocapsules de polymère(D). |
| Dans le texte | |
|
Figure 2. Le passage des systèmes particulaires à travers l’épithélium intestinal. L’épithélium intestinal est formé d’une monocouche de cellules étroitement liées les unes aux autres par des jonctions serrées. Le principe actif ou le vecteur pharmaceutique peuvent emprunter trois voies de passage à travers cet épithélium afin de quitter la lumière intestinale et rejoindre la circulation sanguine ou lymphatique : la voie paracellulaire (passage entre deux cellules adjacentes), la voie transcellulaire (absorption par les entérocytes, cellules spécialisées dans l’absorption des nutriments) et la voie lymphatique (absorption par les cellules M associées aux follicules lymphoïdes de l’intestin). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.