")
")
| Issue |
Med Sci (Paris)
Volume 23, Number 2, Février 2007
|
|
|---|---|---|
| Page(s) | 161 - 166 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/2007232161 | |
| Published online | 15 février 2007 | |
Anomalies d’expression du complexe récepteur T de l’antigène/CD3 et déficits immunitaires
CD3 complex and immunodeficiencies
1
Département de microbiologie et d’immunologie, Université de Montréal, CHU Sainte-Justine, 3175, cheminde la Côte Sainte-Catherine, Montréal (Québec), H3T 1C5 Canada
2
Inserm U768, Hôpital Necker, 149, rue de Sèvres, 75743 Paris Cedex 15, France
3
Inserm U653, Institut Curie, pavillon Pasteur, section Recherche, 26, rue d’Ulm, 75248 Paris Cedex 15, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
La caractérisation moléculaire des déficits immunitaires contribue à une meilleure compréhension des mécanismes physiologiques du système immunitaire. Le récepteur T de l’antigène est constitué d’un hétérodimère (α/β ou γ/δ) associé à 4 protéines transmembranaires : CD3γ, δ, ε, et ζ. Aujourd’hui, des déficits impliquant les quatre chaînes du CD3 ont été identifiés. Nous discutons ici des déficits immunitaires liés à des anomalies génétiques délétères touchant chacune des chaînes du complexe CD3 et nous comparons les phénotypes observés chez ces patients avec les connaissances apportées par les souris dont les gènes sont invalidés. Ces observations permettent d’illustrer la fonction respective de chacune de ces chaînes dans le développement des lymphocytes T et de souligner les différences entre le développement lymphocytaire humain et murin.
Abstract
Molecular characterization of immunodeficiencies contributes to a better understanding of the physiological mechanisms of immune function. The T cell receptor is a heterodimer (α/β or γ/δ) associated with four transmembrane units of the CD3 complex (γ, δ, ε and ζ). We herein summarize the immunodeficiency states resulting from defects in genes encoding the CD3 complex. Such analysis highlights the respective role of each of these chains in T lymphocyte development and underscores differences between T lymphocyte development in man and mouse. Currently, there is a growing body of knowledge on immunodeficiencies specifically involving the four chains of the CD3, namely γ, δ, ε and ζ. Thus, we can compare the phenotypes observed in these patients with those seen in mice knockout for these genes. The main differences observed involve the respective roles of the CD3γ chain as well as the CD3δ, whose functions seem to be reciprocal between the two species. Indeed, in the mouse, knockout of CD3δ allows some degree of T lymphocyte differentiation since mature CD4 and CD8 as well as TCRγδ T lymphocytes are observed in the periphery. In contrast, deleterious mutation of the CD3δ encoding gene in the human leads to a severe combined immunodeficiency characterised by the complete absence of mature T cell subpopulations including TCRα/β and TCRγ/δ. Reciprocally, in the human, mutation of the CD3γ encoding gene leads to a moderate immunodeficiency which contrasts with the complete block of T cell differentiation observed in mice knockout for this gene. This article brings into focus the knowledge gained through studies of immunodeficiency mouse models with the pathophysiological state observed in human disease.
© 2007 médecine/sciences - Inserm / SRMS
Les déficits immunitaires combinés sévères (DICS) constituent un groupe de maladies rares caractérisées par un arrêt de la différenciation des lymphocytes T. L’identification des causes moléculaires de cet arrêt permet de mieux comprendre les mécanismes de développement des lymphocytes T chez l’homme. Actuellement, les défauts génétiques à l’origine des DICS permettent de distinguer au moins quatre types de mécanismes conduisant à l’arrêt du développement des lymphocytes T [1]. (1) La mort des thymocytes est en partie responsable de la lymphopénie T observée dans le cas du déficit en adénosine désaminase [2]. (2) Les mutations de la chaîne γc [3] ou de la tyrosine kinase JAK-3 associée à γc [4] et de la chaîne α du récepteur de l’interleukine-7 [5] entraînent un défaut de transduction de signal dépendant de certaines interleukines. (3) Les anomalies génétiques de RAG-1, de RAG-2 [6] et d’Artémis [7] expliquent le défaut de réarrangement des gènes codant les chaînes du récepteur de l’antigène. (4) À un déficit de la phosphatase CD45 est attribuable un défaut de transduction de signal par le pré-TCR [8]. Plus récemment, des mutations dans les gènes codant les chaînes du complexe CD3 δ, γ et ζ ont été observées et illustrent aussi ce dernier mécanisme [9, 10]. Au contraire, le phénotype modéré du déficit immunitaire associé à des mutations de CD3γ démontre une hétérogénéité fonctionnelle des chaînes du CD3 au sein du complexe TCR/CD3.
Le complexe TCR/CD3
Contrairement aux récepteurs à activité tyrosine kinase comme le récepteur de l’insuline qui présente la double fonction de reconnaissance et de transduction de signal, le complexe TCR/CD3 dissocie ces deux fonctions. La reconnaissance de l’antigène est assurée par le récepteur T de l’antigène (TCR), hétérodimère αβ ou γδ, alors que le complexe CD3 en recrutant des tyrosine kinases de la famille Src et Syk [11] transmet un signal intracytoplasmique. Les chaînes CD3γ, CD3δ, CD3ε et CD3ζ forment le complexe CD3 sous forme de modules dimériques, δε, γε et ζζ (Figure 1). Selon ce modèle de stochiométrie [12], le complexe CD3 contient au total dans sa partie intracytoplasmique, dix motifs nommés ITAM (immunoreceptor tyrosine-based activation motif), contenant chacun deux résidus tyrosine séparés par 9 à 12 acides aminés. Trois sont portés par CD3ζ, et un par chacune des autres chaînes : CD3γ, CD3δ et CD3ε. La phosphorylation de ces motifs par des protéine kinases de la famille Src conduit au recrutement de la tyrosine kinase appartenant à la famille Syk, ZAP70 (zeta associated kinase), qui associée à CD3ζ, amorce l’activation des lymphocytes T.
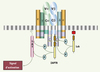 |
Figure 1. Le complexe TCR/CD3. Le récepteur T de l’antigène est constitué d’un hétérodimère (α/β ou γ/δ) associé à 4 protéines transmembranaires : CD3γ, δ, ε, et ζ. Les chaînes CD3γ, CD3δ, CD3ε et CD3ζ forment des modules dimériques δε, γε et ζζ. Chaque chaîne γ, δ, ε, et ζ présente dans sa partie intracytoplasmique des motifs ITAM (en violet) qui sont phosphorylés sur les résidus tyrosine par la tyrosine kinase Lck. La phosphorylation des ITAM de ζ permet l’association de ZAP70 à cette chaîne par l’intermédiaire des 2 domaines SH2 (en marron) portés par cette kinase et secondairement sa phosphorylation par Lck. ZAP70 ainsi activée phosphoryle à son tour l’adaptateur LAT. |
Le développement intrathymique
L’expression des molécules CD4 et CD8 définit trois stades de maturation des thymocytes. Les thymocytes « double négatif » CD4-CD8- progressent vers le stade « double positif » CD4+CD8+ avant de se différencier en thymocytes simple positif CD4+ ou CD8+ qui quittent le thymus sous forme de lymphocytes T matures. Le réarrangement des gènes codant la chaîne β du TCR se produit au stade « double négatif » et précède le réarrangement des gènes codant la chaîne α. La transition du stade « double négatif » au stade « double positif » nécessite l’expression membranaire du pré-TCR, constitué de la chaîne TCRβ associée à la chaîne invariante pré-Tα, auquel est associé le complexe CD3. La transition des thymocytes « double positif » en thymocytes « simple positif » CD4+ ou CD8+ dépend de l’expression du TCRα/β. L’expression successive du pré-TCR puis du TCR induit des signaux intenses de division cellulaire. L’expression du TCR par les thymocytes « double positif » permet la sélection positive des thymocytes. Les souris dont les différentes chaînes du complexe CD3 sont invalidées illustrent le rôle respectif de ces protéines au cours de la différenciation. Ainsi l’absence de la chaîne CD3ε ou CD3γ [13] conduit à un arrêt de maturation des thymocytes au stade « double négatif » tandis que l’invalidation du gène CD3δ conduit à un arrêt au stade double positif et épargne les thymocytes portant un TCRγδ [14]. Cela démontre que le module dimérique γε est essentiel à un stade plus précoce que le module δε dans le développement des thymocytes. La chaîne CD3ζ est indispensable à la transition du stade « double négatif »-« simple positif » [15]. Cependant, le phénotype des déficits immunitaires liés à une anomalie d’une de ces chaînes montre clairement que leur rôle respectif dans la différenciation intrathymique chez l’homme est différent de celui observé chez la souris.
Déficit de la chaîne CD3δ
Des mutations du gène CD3D ont été détectées chez 8 enfants de 4 familles présentant un DICS avec absence totale de lymphocytes T et présence normale de lymphocytes B et NK (DICS T- B+ NK+)[9, 10, 16]. Dans une famille, le diagnostic de DICS porté chez deux fœtus a conduit à des interruptions de grossesse [10]. Dans tous ces cas, les mutations décrites ont été observées à l’état homozygote, conséquence de la consanguinité des parents clairement démontrée dans au moins 3 familles [9, 10]. Une mutation identique a été retrouvée dans deux familles d’origine différente (marocaine dans un cas et mennonite dans l’autre) [9, 10].
Deux mutations entraînent des codons stop prématurés et donc des protéines tronquées dans leur partie extracellulaire [9, 10]. La troisième mutation touche le site accepteur d’épissage de l’intron 2 et conduit à une anomalie d’épissage caractérisée par un ARN dépourvu de l’exon 3 codant principalement la partie transmembranaire de la protéine [16] (Figure 2A). Ainsi, les mutations de CD3D décrites chez des patients atteints de DICS T- B+ NK+ entraînent la synthèse de protéines sans région transmembranaire donc incapables de s’associer avec CD3ε et, de ce fait, non exprimées au niveau de la membrane. L’absence de lymphocytes T est totale chez ces patients et est associée à la survenue d’infections graves dès les premiers mois de la vie. L’absence de lymphocytes T est confirmée dans les ganglions lymphatiques fœtaux.
 |
Figure 2. Mutations des gènes codant les différentes chaînes du CD3 et protéines produites. Les mutations touchant les gènes des différentes chaînes du CD3γ, δ, ε, et ζ actuellement décrites sont visualisées. Les protéines produites sont comparées aux protéines normales. EC : extracellulaire ; IC : intracellulaire ; TM : transmembranaire. |
L’analyse de biopsies thymiques d’un patient par étude d’expression de transcrits et de protéines [9] ou, chez des fœtus, par étude immuno-histochimique [10], apporte des informations sur le stade d’arrêt de la différenciation des thymocytes. Dans le thymus du patient, la faible expression de CD4 et de CD8 (chaînes α et β) et des chaînes TCRα et TCRβ suggèrent un blocage de différenciation au stade double négatif. Les thymus fœtaux sont caractérisés par une faible cellularité, une faible prolifération des thymocytes et par la présence de thymocytes ISP (intermediate single positive), qui correspondent à un stade intermédiaire entre le stade « double négatif » et « double positif » caractérisé par l’expression de CD45RO et de CD4, ainsi que la perte d’expression de CD34 [17]. Ces deux études démontrent un arrêt précoce dans la différenciation des thymocytes avec un blocage de la différenciation au moment de la transition entre stade « double négatif » et stade « double positif ».
Cette observation montre le caractère indispensable de CD3δ dans la signalisation par le pré-TCR chez l’homme. En revanche, l’invalidation du gène CD3δ chez la souris entraîne un blocage au stade « double positif » [14]. À ce stade, CD3δ pourrait induire l’activation, lors de la sélection positive, de la voie d’activation dépendante des kinases ERK [18]. Cet arrêt de la différenciation est donc plus tardif que chez l’homme. Il n’est, de plus, pas complet puisque l’on détecte dans la rate de ces souris des lymphocytes T matures CD4+ et CD8+ exprimant un nombre réduit de complexes CD3/TCR au niveau de la membrane. En outre, la différenciation des lymphocytes portant un TCRγδ qui sont en quantité normale est épargnée. Cela contraste avec l’absence totale de lymphocytes T TCRα/β et γ/δ chez les patients (Figure 3). Il n’y a pas, à ce jour, d’explication satisfaisante qui permet de comprendre les fonctions distinctes de CD3δ chez l’homme et la souris, bien que la structure de CD3δ soit très conservée.
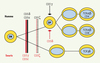 |
Figure 3. Rôle des différentes chaînes du complexe CD3 dans la différenciation lymphocytaire T chez l’homme et chez la souris. Le défaut des différentes chaînes du CD3 entraîne des blocages, soit complets, soit partiels, de la différenciation lymphocytaire T chez l’homme et chez la souris. Le rôle respectif des différentes chaînes, en particulier de γ et de δ, est différent chez l’homme et chez la souris. |
Déficit de la chaîne CD3ε
Dans une famille consanguine, un déficit complet de CD3ε a été récemment décrit chez deux enfants atteints d’un DICS T -B+NK+. La mutation observée sous forme homozygote chez les patients consiste en une délétion de deux paires de bases à la position 1289 de l’exon 5 du gène CD3E conduisant à un décalage du cadre de lecture et à un codon stop prématuré et donc à une protéine tronquée dans sa partie extracytoplasmique (Figure 2B). En l’absence de tissu thymique provenant de ces patients, nous ne pouvons connaître précisément le stade de blocage de la différenciation thymocytaire. Dans les modèles murins, l’invalidation de la chaîne CD3ε entraîne un blocage au stade double négatif, blocage secondaire à un défaut de signal et de prolifération en aval du récepteur pré-TCR [19, 20]. Nous pouvons supposer que la chaîne CD3ε joue un rôle analogue chez l’homme (Figure 3) compte tenu de la participation de CD3ε dans les hétérodimères CD3γε et CD3δε.
Une forme partielle de déficit de la chaîne CD3ε avait été décrite antérieurement chez un patient qui présentait un déficit immunitaire partiel. Les lymphocytes T de ce patient se caractérisent par une expression membranaire réduite du complexe TCR/CD3 (environ 10 % de la normale) contrastant avec un phénotype normal. Cela suggère qu’une expression normale du complexe TCR/CD3 n’est pas indispensable à une différenciation thymique normale. Ce patient présente une double mutation hétérozygote. La première, héritée de son père, est une mutation qui touche l’exon 6 et qui entraîne un codon stop prématuré dans la partie extracytoplasmique de la protéine. La deuxième, héritée de sa mère, est une mutation qui touche le site d’épissage donneur de l’intron 7 conduisant à un ARN dépourvu de l’exon 7 et donc à une protéine dépourvue de région transmembranaire. Cependant, un certain degré d’épissage normal persiste et explique l’expression résiduelle du complexe TCR/CD3 détectée à la membrane des lymphocytes T de ce patient [21] (Figure 2B).
Déficit de la chaîne CD3γ
Un déficit de la chaîne CD3γ a été détecté chez trois patients de deux familles, l’une espagnole et l’autre turque [22–24]. Le tableau clinique observé est très variable d’un patient à l’autre, même au sein d’une même famille. En effet, l’un des patients est décédé à l’âge de 32 mois des suites d’infections associées à une anémie hémolytique et à une entéropathie auto-immune grave. Son frère, à l’âge de 18 ans, ne présente que des manifestations auto-immunes. Le troisième enfant, après avoir souffert d’infections respiratoires dans la petite enfance, est, à l’âge de 7 ans, en bonne santé sous traitement prophylactique par immunoglobulines intraveineuses.
Les lymphocytes T de ces patients sont caractérisés par une faible expression membranaire du complexe TCR/CD3 de l’ordre de 20 % à 30 % par rapport à la normale. Par ailleurs, une lymphopénie CD8 existe chez deux patients. Le nombre réduit de lymphocytes T naïfs CD45RA+ suggère un défaut de la thymopoïèse.
Les deux patients de la famille espagnole présentent une double mutation hétérozygote. La mutation héritée du père transforme le codon initiateur, ATG, en codon GTG et donc aucune protéine ne peut être produite. La mutation héritée de la mère affecte le site accepteur d’épissage de l’intron 2 entraînant une perte de 17 nucléotides dans l’ARN et un codon stop prématuré [22]. Les analyses biochimiques montrent que cette protéine, tronquée dans sa partie extracytoplasmique, n’est pas exprimée à la membrane des lymphocytes. Le patient turc, dont les parents sont consanguins, présente une mutation homozygote qui consiste en une substitution d’une base dans l’exon 3 conduisant à un codon stop prématuré et à une protéine tronquée dans sa partie extracytoplasmique (Figure 2C) [23].
Le phénotype modéré du déficit immunitaire de ces patients contraste avec le blocage au stade thymocyte « double négatif », secondaire au défaut de transduction de signal par le pré-TCR observé chez la souris dont le gène CD3G a été invalidé (Figure 3). Chez la souris, il n’existe aucune possibilité d’expression d’un complexe TCR/CD3 en l’absence de CD3γ contrairement à l’homme [25]. Cela suggère que le remplacement de la chaîne CD3γ par la chaîne CD3δ qui corrige la fonction du complexe TCR/CD3 dans des cellules humaines [25], ne se fait pas chez la souris.
Déficit de la chaîne CD3ε
Récemment, nous avons décrit chez un patient présentant une susceptibilité accrue aux infections bactériennes, virales et fungiques, un déficit de CD3ζ lié à une mutation homozygote du gène CD3Z [26]. Chez ce patient, l’analyse phénotypique est très particulière : elle montre l’existence de deux populations lymphocytaires T, l’une (90 % des lymphocytes T) exprime faiblement le complexe TCR/CD3 à la membrane, l’autre l’exprime normalement. Dans la première population, l’expression résiduelle est très faible, environ 100 fois moins que la normale et le complexe exprimé est dépourvu de chaîne CD3ζ. Cependant, cette population se répartit normalement en lymphocytes T CD4+ et CD8+ mais est principalement de phénotype mémoire CD45RO. Cela montre qu’en l’absence de chaîne CD3ζ, l’expression du complexe TCR/CD3 n’est pas complètement abolie. Cette expression résiduelle permet une différenciation des lymphocytes T. Cette population lymphocytaire présente une mutation homozygote du gène CD3Z en position 207 (C → T) avec pour conséquence un codon stop en position 70, c’est-à-dire 2 acides aminés avant le premier résidu tyrosine du premier motif ITAM (Figure 2D). Cette mutation induit la synthèse d’une protéine dépourvue des trois motifs ITAM, et cette protéine n’est pas exprimée à la membrane (Figure 2D). La souris dont la chaîne CD3ζ est invalidée présente un phénotype analogue. Le blocage de la différenciation thymocytaire entre le stade « double négatif » et le stade « double positif » n’est pas total puisque des lymphocytes T matures sont détectés en périphérie et s’accumulent au cours du temps (Figure 3) [15, 27]. Ces lymphocytes expriment faiblement le complexe TCR/CD3 et présentent un défaut de l’activation dépendante du TCR. Leur activité potentiellement auto-réactive démontre le rôle primordial de l’homodimère ζζ au cours de la sélection négative [28].
Chez le patient, dans la population exprimant normalement le complexe TCR/CD3, entièrement constituée de lymphocytes CD4+, trois séquences différentes de CD3Z ont été détectées. Ces séquences sont caractérisées par l’association de la transition du nucléotide 207 à une autre mutation au niveau du nucléotide 208 ou 209 transformant ainsi le codon stop en une substitution d’acide aminé en position 70 permettant, de ce fait, l’expression d’une protéine de poids moléculaire normal à la membrane (Figure 2D). Ces trois séquences correspondent à des mutations somatiques qui se sont produites à partir de la mutation germinale constituée par la transition C → T à la position 207. Ce résultat suggère que les mutations somatiques sont beaucoup plus fréquentes que ce que l’on pouvait envisager. Cette observation met en exergue l’avantage sélectif considérable que procure la survenue de mutations partiellement correctrices comme celles déjà observées dans d’autres déficits immunitaires touchant les lymphocytes T [29–32]. Par analogie au défaut de ZAP70 caractérisé chez l’homme par l’absence de CD8 [33], l’absence de lymphocytes T CD8 au sein de cette population reflète probablement le défaut de signalisation dépendante de ZAP70 observé dans ces cellules.
Conclusions
L’identification de défauts dans une des chaînes du CD3 responsables de déficits immunitaires souligne la diversité des causes moléculaires de ce groupe de maladies. Il est probable que ce type de défauts est plus fréquent qu’on ne le pensait et que nous en comprendrons beaucoup mieux les mécanismes par l’examen et la description de nouveaux cas.
Les conséquences phénotypiques de ces défauts élucident, au moins en partie, le rôle respectif de chaque chaîne du complexe CD3 dans la différenciation et l’activation des lymphocytes T chez l’homme ; de plus, elles soulignent les différences entre l’homme et les modèles murins.
Article reçu le 28 mars 2006, accepté le 16 juin 2006.
Références
- Fischer A, Le Deist F, Hacein-Bey-Abina S, et al. Severe combined immunodeficiency. A model disease for molecular immunology and therapy. Immunol Rev 2005; 203 : 98–109. [Google Scholar]
- Hirschhorn R. Immunodeficiency disease due to deficiency of adenosine deaminase. In: Ochs HD, Smith CIE, Puck JM, eds. Primary immunodeficiency diseases. A molecular and genetic approach. New York : Oxford University Press, 1999 : 121–39. [Google Scholar]
- Leonard WJ, Noguchi M, Russell SM, McBride OW. The molecular basis of X-linked severe combined immunodeficiency: the role of the interleukin-2 receptor gamma chain as a common gamma chain, gamma c. Immunol Rev 1994; 138 : 61–86. [Google Scholar]
- Macchi P, Villa A, Giliani S, et al. Mutations of Jak-3 gene in patients with autosomal severe combined immune deficiency (SCID). Nature 1995; 377 : 65–8. [Google Scholar]
- Puel A, Ziegler SF, Buckley RH, Leonard WJ. Defective IL7R expression in T(-)B(+)NK(+) severe combined immunodeficiency. Nat Genet 1998; 20 : 394–7. [Google Scholar]
- Schwarz K, Gauss GH, Ludwig L, et al. RAG mutations in human B cell-negative SCID. Science 1996; 274 : 97–9. [Google Scholar]
- Moshous D, Callebaut I, de Chasseval R, et al. Artemis, a novel DNA double-strand break repair/V(D)J recombination protein, is mutated in human severe combined immune deficiency. Cell 2001; 105 : 177–86. [Google Scholar]
- Kung C, Pingel JT, Heikinheimo M, et al. Mutations in the tyrosine phosphatase CD45 gene in a child with severe combined immunodeficiency disease. Nat Med 2000; 6 : 343–5. [Google Scholar]
- Dadi HK, Simon AJ, Roifman CM. Effect of CD3delta deficiency on maturation of alpha/beta and gamma/delta T-cell lineages in severe combined immunodeficiency. N Engl J Med 2003; 349 : 1821–8. [Google Scholar]
- De Saint Basile G, Geissmann F, Flori E, et al. Severe combined immunodeficiency caused by deficiency in either the delta or the epsilon subunit of CD3. J Clin Invest 2004; 114 : 1512–7. [Google Scholar]
- Werlen G, Palmer E. The T-cell receptor signalosome: a dynamic structure with expanding complexity. Curr Opin Immunol 2002; 14 : 299–305. [Google Scholar]
- Call ME, Wucherpfennig KW. Molecular mechanisms for the assembly of the T cell receptor-CD3 complex. Mol Immunol 2004; 40 : 1295–305. [Google Scholar]
- Haks MC, Krimpenfort P, Borst J, Kruisbeek AM. The CD3gamma chain is essential for development of both the TCRalphabeta and TCRgammadelta lineages. EMBO J 1998; 17 : 1871–82. [Google Scholar]
- Dave VP, Cao Z, Browne C, et al. CD3 delta deficiency arrests development of the alpha beta but not the gamma delta T cell lineage. EMBO J 1997; 16 : 1360–70. [Google Scholar]
- Love PE, Shores EW, Johnson MD, et al. T cell development in mice that lack the zeta chain of the T cell antigen receptor complex. Science 1993; 261 : 918–21. [Google Scholar]
- Takada H, Nomura A, Roifman CM, Hara T. Severe combined immunodeficiency caused by a splicing abnormality of the CD3delta gene. Eur J Pediatr 2005; 164 : 311–4. [Google Scholar]
- Fukuhara K, Okumura M, Shiono H, et al. A study on CD45 isoform expression during T-cell development and selection events in the human thymus. Hum Immunol 2002; 63 : 394–404. [Google Scholar]
- Delgado P, Fernandez E, Dave V, et al. CD3delta couples T-cell receptor signalling to ERK activation and thymocyte positive selection. Nature 2000; 406 : 426–30. [Google Scholar]
- Malissen M, Gillet A, Ardouin L, et al. Altered T cell development in mice with a targeted mutation of the CD3-epsilon gene. EMBO J 1995; 14 : 4641–53. [Google Scholar]
- Sommers CL, Dejarnette JB, Huang K, et al. Function of CD3 epsilon-mediated signals in T cell development. J Exp Med 2000; 192 : 913–19. [Google Scholar]
- Soudais C, de Villartay JP, Le Deist F, et al. Independent mutations of the human CD3-epsilon gene resulting in a T cell receptor/CD3 complex immunodeficiency. Nat Genet 1993; 3 : 77–81. [Google Scholar]
- Arnaiz-Villena A, Timon M, Corell A, et al. Brief report: primary immunodeficiency caused by mutations in the gene encoding the CD3-gamma subunit of the T-lymphocyte receptor. N Engl J Med 1992; 327 : 529–33. [Google Scholar]
- Regueiro JR, Pacheco A, Alvarez-Zapata D, et al. CD3 deficiencies. In: Ochs HD, Smith CIE, Puck JM, eds. Primary immunodeficiencies diseases: a moleclular and genetic approach. New York : Oxford University Press, 1999 : 189–97. [Google Scholar]
- Van Tol M, Sanal O, Langlois Van den Bergh R, et al. CD3γ chain deficiency leads to a cellular immunodeficiency with a mild clinical presentation. Immmunologist 1997; (suppl 1): 41–3. [Google Scholar]
- Torres PS, Alcover A, Zapata DA, et al. TCR dynamics in human mature T lymphocytes lacking CD3 gamma. J Immunol 2003; 170 : 5947–55. [Google Scholar]
- Rieux-Laucat F, Hivroz C, Lim A, et al. Inherited and somatic CD3zeta mutations in a patient with T-cell deficiency. N Engl J Med 2006; 354 : 1913–21. [Google Scholar]
- Malissen M, Gillet A, Rocha B, et al. T cell development in mice lacking the CD3-zeta/eta gene. EMBO J 1993; 12 : 4347–55. [Google Scholar]
- Lin SY, Ardouin L, Gillet A, et al. The single positive T cells found in CD3-zeta/eta−/− mice overtly react with self-major histocompatibility complex molecules upon restoration of normal surface density of T cell receptor-CD3 complex. J Exp Med 1997; 185 : 707–15. [Google Scholar]
- Stephan V, Wahn V, Le Deist F, et al. Atypical X-linked severe combined immunodeficiency due to possible spontaneous reversion of the genetic defect in T cells. N Engl J Med 1996; 335 : 1563–7. [Google Scholar]
- Ariga T, Yamada M, Sakiyama Y, Tatsuzawa O. A case of Wiskott-Aldrich syndrome with dual mutations in exon 10 of the WASP gene: an additional de novo one-base insertion, which restores frame shift due to an inherent one-base deletion, detected in the major population of the patient’s peripheral blood lymphocytes. Blood 1998; 92 : 699–701. [Google Scholar]
- Wada T, Toma T, Okamoto H, et al. Oligoclonal expansion of T lymphocytes with multiple second-site mutations leads to Omenn syndrome in a patient with RAG1-deficient severe combined immunodeficiency. Blood 2005; 106 : 2099–101. [Google Scholar]
- Hirschhorn R, Yang DR, Puck JM, et al. Spontaneous in vivo reversion to normal of an inherited mutation in a patient with adenosine deaminase deficiency. Nat Genet 1996; 13 : 290–5. [Google Scholar]
- Arpaia E, Shahar M, Dadi H, et al. Defective T cell receptor signaling and CD8+ thymic selection in humans lacking zap-70 kinase. Cell 1994; 76 : 947–58. [Google Scholar]
Liste des figures
|
Figure 1. Le complexe TCR/CD3. Le récepteur T de l’antigène est constitué d’un hétérodimère (α/β ou γ/δ) associé à 4 protéines transmembranaires : CD3γ, δ, ε, et ζ. Les chaînes CD3γ, CD3δ, CD3ε et CD3ζ forment des modules dimériques δε, γε et ζζ. Chaque chaîne γ, δ, ε, et ζ présente dans sa partie intracytoplasmique des motifs ITAM (en violet) qui sont phosphorylés sur les résidus tyrosine par la tyrosine kinase Lck. La phosphorylation des ITAM de ζ permet l’association de ZAP70 à cette chaîne par l’intermédiaire des 2 domaines SH2 (en marron) portés par cette kinase et secondairement sa phosphorylation par Lck. ZAP70 ainsi activée phosphoryle à son tour l’adaptateur LAT. |
| Dans le texte | |
|
Figure 2. Mutations des gènes codant les différentes chaînes du CD3 et protéines produites. Les mutations touchant les gènes des différentes chaînes du CD3γ, δ, ε, et ζ actuellement décrites sont visualisées. Les protéines produites sont comparées aux protéines normales. EC : extracellulaire ; IC : intracellulaire ; TM : transmembranaire. |
| Dans le texte | |
|
Figure 3. Rôle des différentes chaînes du complexe CD3 dans la différenciation lymphocytaire T chez l’homme et chez la souris. Le défaut des différentes chaînes du CD3 entraîne des blocages, soit complets, soit partiels, de la différenciation lymphocytaire T chez l’homme et chez la souris. Le rôle respectif des différentes chaînes, en particulier de γ et de δ, est différent chez l’homme et chez la souris. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.