")
")
| Issue |
Med Sci (Paris)
Volume 21, Number 2, Février 2005
|
|
|---|---|---|
| Page(s) | 132 - 133 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/2005212132 | |
| Published online | 15 février 2005 | |
Les exosomes : des convoyeurs de prions ?
Exosomes: Carriers of prions ?
1
UMR144-CNRS, Institut Curie, 26, rue d’Ulm, 75248 Paris Cedex 05, France
2
Inra, Unité de Virologie et immunologie moléculaires, 78350 Jouy-en-Josas, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
*
vilette@ jouy.inra.fr
Peu étudiées jusqu’au récent épisode de l’encéphalopathie spongiforme bovine, les maladies à prions - ou encore encéphalopathies spongiformes transmissibles - sont des maladies neurodégénératives à l’issue invariablement fatale qui touchent aussi bien l’homme (maladie de Creutzfeldt-Jakob) que l’animal [1]. La conversion, dans les tissus infectés, d’une protéine cellulaire, la PrP, en une isoforme conformationnelle anormale et pathologique est un événement central de ces affections. La PrP anormale (ou PrPSc) serait non seulement à l’origine de la neurodégénérescence [2] et des dysfonctionnements neurologiques observés dans ces maladies mais aussi, selon l’hypothèse dite « de la protéine seule », l’agent infectieux [3]. La conversion de la PrP en PrPSc, via une interaction des deux isoformes, constituerait ainsi le mode de multiplication de ces agents infectieux non conventionnels.
Si les lésions dégénératives observées en phase terminale sont restreintes au système nerveux central, ce tissu ne constitue pas pour autant le siège exclusif de la multiplication des prions. Dans la grande majorité des cas, les contaminations ont en effet lieu par voie périphérique, le plus souvent par voie orale. Après franchissement de la barrière intestinale, l’agent infectieux se multiplie dans diverses formations lymphoïdes (ganglions, rate…), puis colonise le système nerveux central via les trajets nerveux qui relient les organes lymphoïdes à la moelle épinière [4]. Des trajets alternatifs, impliquant les fibres sympathiques qui innervent la paroi intestinale, permettent cependant aux prions d’envahir le cerveau sans multiplication préalable dans les tissus lymphoïdes. L’identification de certains des tissus périphériques assurant la multiplication et/ou le transport des prions vers l’organe cible - le cerveau - est à l’évidence essentielle à la compréhension de la pathogénie de ces maladies [5]. Cependant - et en raison de la nature très particulière de ces agents - les mécanismes précis mis en jeu pour assurer leur dissémination dans l’organisme restent très mal compris. Une fois les cellules infectées, la forme normale de la PrP ainsi que son isoforme anormale sont présentes à la surface cellulaire [6]. Dès lors, un contact membranaire est-il suffisant pour qu’une cellule s’infecte au contact d’une cellule infectée ? Peut-on, au contraire, imaginer que des prions soient sécrétés et qu’ils infectent des cellules cibles éloignées ? Ces deux cas de figure peuvent-ils coexister ?
Des travaux publiés récemment font état de formes libres de l’agent infectieux, sécrétées par des cellules infectées [7]. En utilisant des cultures de cellules infectables par les prions (lignées Rov et Mov), nous avons montré que ces cultures excrètent la forme normale de la PrP cellulaire, quand elles ne sont pas infectées, et la forme anormale PrPsc après infection par une souche de prions ovins. Le fractionnement par ultracentrifugation à 100 000 g du milieu conditionné nous a permis de montrer que les deux isoformes de la PrP sont associées à des petites vésicules membranaires qui, sur la base de leur morphologie, de leur taille (50 à 90 nm), de leur composition protéique et lipidique et de leur densité, possèdent les caractéristiques des exosomes. Les exosomes sont de très petites vésicules d’origine endosomique sécrétées par certaines cellules lors de la fusion des endosomes tardifs multivésiculaires avec la membrane plasmique [8]. L’inoculation de ces vésicules exosomiques à l’animal confirme la présence de prions infectieux associés à ces vésicules. Ces travaux - en accord avec des observations plus anciennes localisant la PrPsc dans les endosomes tardifs et les lysosomes des cellules infectées [6] - montrent que la fusion de compartiments intracellulaires avec la membrane plasmique permet la sécrétion de prions infectieux associés aux membranes exosomiques. Ces travaux suggèrent donc que les exosomes - vecteurs potentiels de communication intercellulaire - participent à la dissémination des prions dans l’organisme. L’hypothèse d’une infection à distance soulevée par ce travail pourrait rendre compte de la progression de l’infection en cas de discontinuité physique des partenaires cellulaires permissifs (Figure 1). Pour autant, la mise en évidence de formes libres de l’agent infectieux n’exclut pas d’autres modes de transfert des prions. La présence de PrP anormale à la surface des cellules infectées pourrait, par contacts de membrane à membrane, déclencher les premiers événements de conversion à la surface des cellules cibles. L’infection pourrait alors progresser de proche en proche, entre cellules adjacentes, et il n’est d’ailleurs pas exclu que les exosomes puissent également jouer un rôle dans un tel processus.
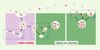 |
Figure 1. Mécanismes possibles du transfert intercellulaire des exosomes. Les exosomes correspondent aux vésicules intraluminales des endosomes. Ces vésicules sont produites au cours de la maturation des endosomes (1). Dans la cellule infectée (en violet), la PrPSc (rose foncé) est portée par les vésicules au sein des cellules. Ces vésicules ont la possibilité d’être sécrétées dans le milieu extracellulaire et sont alors appelées « exosomes ». Ces exosomes peuvent transmettre la PrPSc à la cellule voisine (2), ou à distance (3). Les cellules non infectées (en vert) vont recevoir la PrPsc par un processus qui reste à démontrer : fusion de la vésicule avec la surface cellulaire, endocytose de vésicule entière ou phagocytose de plusieurs vésicules. |
Les mécanismes impliqués dans la séquestration de la PrPSc dans les exosomes - ceux qui contrôlent la fusion des endosomes multivésiculaires avec la membrane plasmique et le mode de transfert des exosomes vers d’autres cellules - sont autant de champs d’investigation qui restent à explorer. À terme, ces études pourraient permettre de mieux appréhender les mécanismes participant à la propagation des prions.
Références
- Collinge J. Prion diseases of humans and animals : their causes and molecular basis. Annu Rev Neurosci 2001; 24 : 519–50. [Google Scholar]
- Chiesa R, Harris DA. Prion diseases : what is the neurotoxic molecule ? Neurobiol Dis 2001; 8 : 743–63. [Google Scholar]
- Prusiner SB. Molecular biology of prion diseases. Science 1991; 252 : 1515–22. [Google Scholar]
- Aguzzi A, Heppner FL, Heikenwalder M, et al. Immune system and peripheral nerves in propagation of prions to CNS. Br Med Bull 2003; 66 : 141–59. [Google Scholar]
- Aguzzi A. Prions and the immune system : a journey through gut, spleen, and nerves. Adv Immunol 2003; 81 : 123–71. [Google Scholar]
- Harris DA. Cellular biology of prion diseases. Clin Microbiol Rev 1999; 12 : 429–44. [Google Scholar]
- Fevrier B, Vilette D, Archer F, et al. Cells release prions in association with exosomes. Proc Natl Acad Sci USA 2004; 101 : 9683–8. [Google Scholar]
- Fevrier B, Raposo G. Exosomes : endosomal-derived vesicles shipping extracellular messages. Curr Opin Cell Biol 2004; 16 : 415–21. [Google Scholar]
© 2005 médecine/sciences - Inserm / SRMS
Liste des figures
|
Figure 1. Mécanismes possibles du transfert intercellulaire des exosomes. Les exosomes correspondent aux vésicules intraluminales des endosomes. Ces vésicules sont produites au cours de la maturation des endosomes (1). Dans la cellule infectée (en violet), la PrPSc (rose foncé) est portée par les vésicules au sein des cellules. Ces vésicules ont la possibilité d’être sécrétées dans le milieu extracellulaire et sont alors appelées « exosomes ». Ces exosomes peuvent transmettre la PrPSc à la cellule voisine (2), ou à distance (3). Les cellules non infectées (en vert) vont recevoir la PrPsc par un processus qui reste à démontrer : fusion de la vésicule avec la surface cellulaire, endocytose de vésicule entière ou phagocytose de plusieurs vésicules. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.