")
")
| Issue |
Med Sci (Paris)
Volume 20, Number 5, Mai 2004
|
|
|---|---|---|
| Page(s) | 551 - 556 | |
| Section | SYNTHÈSE | |
| DOI | https://doi.org/10.1051/medsci/2004205551 | |
| Published online | 15 mai 2004 | |
Bases cellulaires de la mécanotransduction dans la cellule endothéliale
Shear and signal transduction in the endothelial cell
Inserm U.541, Hôpital Lariboisière, 41, boulevard de la Chapelle, 75010 Paris, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Les vaisseaux sanguins sont en permanence soumis à des contraintes mécaniques associées à la pression artérielle, de caractère pulsé, et au flux sanguin. Toute variation de ces contraintes est perçue au niveau des cellules vasculaires et se traduit par des modifications fonctionnelles et structurales des vaisseaux. De nombreux récepteurs, présents à la surface des cellules endothéliales, sont sensibles aux forces de cisaillement. Les intégrines, reliant la matrice extracellulaire aux sites d’adhérence focale et au cytosquelette, peuvent transmettre et moduler la tension mécanique dans la cellule. Par ailleurs, les contraintes mécaniques agissent sur les canaux ioniques, stimulant des récepteurs membranaires et induisent des cascades complexes d’événements biochimiques. De nombreuses voies intracellulaires, telles que la voie des MAP-kinases, sont actives par le flux sanguin et aboutissent à l’induction de facteurs de transcription qui contrôlent l’expression des gènes. Ainsi, par des mécanismes purement locaux, le vaisseau sanguin est capable de s’adapter à son environnement mécanique.
Abstract
Blood vessels are permanently subjected to mechanical forces in the form of stretch and shear stress. Any alterations in the hemodynamic environment invariably produce transformations in the vessel wall that will aim to accommodate the new conditions and to ultimately restore basal levels of mechanical forces. Many receptors, present on the surface of endothelial cells, allow vessels to detect subtle changes in shear stress. Inside the cells, cytoskeletal proteins transmit and modulate the tension between integrins, focal adhesion sites, and the extracellular matrix. Besides inducing structural modifications, mechanical forces lead to changes in the ionic composition of cells, mediated by ion channels, stimulate various membrane receptors, and induce complex biochemical cascades. Many intracellular pathways such as the MAP kinase cascade are activated by shear stress and initiate via sequential phosphorylations the activation of transcription factors and subsequent gene expression. Thus, by purely local mechanisms, blood vessels are capable of true autonomic regulation which enables them to adapt to their mechanical environment.
© 2004 médecine/sciences - Inserm / SRMS
Dans les conditions physiologiques, la contrainte de cisaillement 1 moyenne à laquelle est soumise la surface endoluminale vasculaire est remarquablement constante et proche de 10-15 dyn/cm2, quelle que soit la partie du réseau artériel considérée. Cette régularité est assurée par des mécanismes humoraux autocrines et/ou paracrines qui permettent aux vaisseaux sanguins de réagir immédiatement à des modifications hémodynamiques locales. En revanche, si les variations de tonus vasomoteur sont insuffisantes pour compenser les modifications des contraintes mécaniques, les vaisseaux s’adaptent à leur nouvel environnement hémodynamique en ajustant leur calibre et la structure de leur paroi pour normaliser la contrainte de cisaillement imposée à la surface endothéliale [1, 2]. L’augmentation chronique du débit sanguin stimule la production et l’activation de métalloprotéases matricielles (MMP) qui dégradent les fibres élastiques et contribuent directement à la distension du vaisseau [3]. Cet élargissement du calibre vasculaire étire les cellules musculaires lisses, ce qui entraîne à long terme une hypertrophie de la média (Figure 1). Les contraintes de cisaillement sont également très directement associées à la localisation des plaques d’athérosclérose. Les sites vasculaires susceptibles de développer des plaques d’athérosclérose sont ceux ou le flux sanguin est turbulent ou réversible plutôt que continu et laminaire.
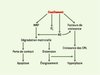 |
Figure 1. Étapes conduisant au remodelage vasculaire induit par la contrainte de cisaillement. La contrainte de cisaillement induit la production de radicaux libres oxygénés (O2‾), la libération de monoxyde d’azote (NO) et la synthèse de métalloprotéases matricielles (MMP) par les cellules endothéliales. L’interaction de NO avec O2‾ produit du peroxynitrite, qui active les MMP. Ces dernières dégradent la matrice extracellulaire, provoquant d’une part l’apoptose des cellules ayant perdu leurs attaches matricielles, et d’autre part l’augmentation du calibre des vaisseaux. Le NO peut aussi, avec le concours des facteurs de croissance stimulés par la contrainte de cisaillement, participer à l’hyperplasie des cellules vasculaires. CML : cellules musculaires lisses. |
Transduction des signaux mécaniques dans les cellules endothéliales
De nombreux récepteurs présents à la surface des cellules endothéliales sont sensibles aux contraintes mécaniques engendrées par le flux. Outre les modifications structurales induites par les contraintes mécaniques, celles-ci déclenchent des cascades complexes d’événements biochimiques qui conduisent à des changements fonctionnels dans la cellule (Figure 2).
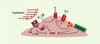 |
Figure 2. Mécanosenseurs dans les cellules endothéliales. Les cellules endothéliales possèdent à leur surface plusieurs récepteurs susceptibles d’être activés par la contrainte de cisaillement, tels que les récepteurs à activité tyrosine kinase (RTyrK), les intégrines (hétérodimères α et β), les canaux potassiques, les protéines G hétérotrimériques (αβγ), et la NAD(P)H oxydase. L’activation d’un ou de plusieurs de ces récepteurs peut conduire à l’entrée de calcium ou à l’activation de différentes cascades biochimiques. Ainsi, l’activation de la NO synthase par la contrainte de cisaillement (et la libération secondaire de monoxyde d’azote [NO]) nécessite une phosphorylation par la protéine kinase Akt (Akt). Shc : protéine adaptatrice Shc ; MAPK : voie des MAP-kinases (mitogen activated protein kinases). |
Intégrines
La matrice extracellulaire participe de façon majeure à la mécanotransduction. Elle renferme des glycoprotéines qui se déplacent sous l’effet du cisaillement et interagissent avec des intégrines spécifiques (αvβ3 avec la fibronectine ou la vitronectine, α6β1 avec la laminine) [4]. Ces dernières, couplées aux protéines du cytosquelette, participent non seulement à l’attachement des cellules à leur substrat, mais aussi à la transmission intracellulaire des signaux mécaniques. Dans des artères coronaires isolées, l’ajout d’un peptide RGD (les protéines de la matrice extracellulaire se lient aux intégrines via la séquence d’acides aminés RGD) ou d’un anticorps anti-intégrine β3 au milieu de culture intraluminal bloque la vasodilatation observée en réponse à l’élévation de flux [5]. Par ailleurs, l’incubation de cellules endothéliales en culture avec un anticorps anti-αvβ3 empêche l’activation du facteur nucléaire NF-қB par la contrainte de cisaillement [6]. La capacité des cellules à détecter la contrainte mécanique, et la réponse biochimique qui en découle, dépendent donc de l’interaction d’intégrines spécifiques avec la matrice extracellulaire.
Canaux ioniques
L’augmentation des contraintes de cisaillement imposées à des cellules endothéliales isolées en culture entraîne une hyperpolarisation de leur membrane cellulaire liée à l’activation de canaux potassiques [7]. L’hyperpolarisation accroît l’entrée du Ca2+, provoquant une accumulation du calcium dans le cytosol et une modification des fonctions cellulaires. Cependant, les mécanismes suscités par les contraintes mécaniques pour contrôler les phénomènes d’ouverture/fermeture de ces canaux sont mal connus. Le plus vraisemblable est que la déformation du cytosquelette induise l’activation des canaux, impliquant une connexion entre ces deux éléments. Une étude confirme d’ailleurs que l’ouverture des canaux K+ est contrôlée par le couplage cytosquelette-protéine G [8]. Par ailleurs, l’hyperpolarisation des cellules sous l’effet de l’augmentation du flux est également sensible aux inhibiteurs des tyrosine kinases [7].
Protéines G
L’activation directe des protéines Gαq/α11 et Gαi3/αo par la contrainte de cisaillement a été rapportée dans les cellules endothéliales [9]. De plus, la régulation de l’expression du proto-oncogène c-fos et de PDGF (platelet-derived growth factor) induite par la contrainte de cisaillement semble dépendre d’un mécanisme PKC-dépendant, nécessitant la présence de Ca2+ et impliquant une protéine G [10]. La sous-unité γ de la protéine G hétérotrimérique est présente au niveau des sites d’adhérence focale riches en intégrines et adjacents aux fibres d’actine-F (stress fibers) [11]. La colocalisation des protéines G et des intégrines permettrait à un même signal d’activer simultanément deux familles de récepteurs transmembranaires (les récepteurs couplés aux protéines G et les intégrines). Par conséquent, les protéines G pourraient être indirectement impliquées dans la voie de signalisation induite par les intégrines.
Récepteurs de type tyrosine kinase
Une autre classe de récepteurs membranaires, de type tyrosine kinase, intervient dans la mécanotransduction. Dans les cellules endothéliales, la contrainte de cisaillement induit la phosphorylation transitoire du récepteur du VEGF (vascular endothelial growth factor) Flk-1 et son association avec la protéine adaptatrice Shc et les intégrines αvβ3 et β1 [12]. Si le rôle de Flk-1 dans la mécanotransduction n’est pas encore parfaitement établi, il n’en reste pas moins que la dissociation de Flk-1 et de Shc et la liaison consécutive de Shc avec Grb2 (growth factor receptor-bound protein 2) et la protéine Sos (son of sevenless), atténue les cascades d’activation en aval ainsi que la transcription de gènes stimulés par la contrainte de cisaillement [12].
Radicaux libres oxygénés
Une part importante de radicaux libres oxygénés produits dans les cellules endothéliales provient de l’oxydase membranaire NADH/NADPH, dont l’activité est réglée par l’écoulement oscillatoire ou laminaire. Cependant, dans le cas des cellules soumises à un flux laminaire, cette activation s’accompagne à long terme d’une augmentation de la superoxyde dismutase qui réduit l’accumulation de radicaux superoxyde O2‾[13], alors que le flux oscillatoire favorise plutôt la surexpression de la sous-unité p22phox de la NAD(P)H [14]. L’accumulation de peroxynitrite, résultant de l’interaction de O2‾avec le monoxyde d’azote (NO), est également stimulée dans un modèle in vivo de fistule artérioveineuse, et participe au remodelage vasculaire par le flux [3] (Figure 1).
Transduction dans la cellule endothéliale
NO et protéine kinase Akt
Un des événements précoces qui survient lorsque les cellules endothéliales en culture sont exposées à un flux laminaire est l’activation de la NO synthase (eNOS) et la libération secondaire de NO. L’activation de eNOS par la contrainte de cisaillement ne requiert pas l’influx de Ca2+ dans la cellule, comme dans le cas d’une activation par des agonistes vasoactifs, mais plutôt une phosphorylation par la protéine kinase Akt [15], elle-même phosphorylée par la phosphatidylinositol-3-kinase (PI3K) [16]. Les voies de signalisation intracellulaire qui font le lien entre la contrainte de cisaillement et l’activation de la eNOS sont nombreuses. D’une part, l’activation de eNOS par la contrainte de cisaillement est inhibée par un bloqueur des canaux potassiques et nécessite un cytosquelette intact. D’autre part, la phosphorylation de eNOS et de Akt dans ces conditions est sensible aux inhibiteurs des tyrosine kinases, indiquant une implication possible de récepteurs tels que ceux au VEGF et à l’insuline [17].
Outre son rôle de vasodilatateur, le NO intervient dans la régulation du remodelage vasculaire induit par la contrainte de cisaillement chronique, puisque l’inhibition de cette voie atténue l’accroissement du calibre des vaisseaux fistulés et restreint l’adaptation à l’augmentation du flux [2]. Dans cette condition, le NO jouerait le rôle de cofacteur, facilitant l’activation des MMP grâce à la production de peroxynitrite [3]. De surcroît, l’activation de Akt et la production de NO favorisent la survie des cellules vasculaires en stimulant des voies anti-apoptotiques et en inhibant des voies pro-apoptotiques (Figure 3) [18].
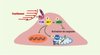 |
Figure 3. Monoxyde d’azote (NO), protéine kinase Akt (Akt) et apoptose. L’activation de la protéine kinase Akt protège la cellule endothéliale de l’apoptose en inhibant la voie de caspases, soit en activant la synthèse de NO par l’intermédiaire de la NO synthase (eNOS), soit en phosphorylant Bad (Bcl-2-antagonist of cell death), empêchant sa liaison avec Bcl-2 (B-cell CLL/lymphoma 2) ou Bcl-XL (Bcl-2-like 1) à la membrane des mitochondries. L’activation d’AKT peut également activer l’expression de gènes favorisant la survie cellulaire. Flk-1 : récepteur à activité tyrosine kinase Flk-1 ; c-Src : kinases c-Src ; PI3K : phosphatidylinositol-3-kinase. |
FAK (focal adhesion kinase)
Au cours de la stimulation des cellules vasculaires par la contrainte de cisaillement, plusieurs événements de signalisation sont associés à la formation des adhérences focales, qui se composent de groupes d’intégrines et de protéines du cytosquelette. Les protéines présentes aux adhérences focales, en particulier la tyrosine kinase cytoplasmique FAK, sont phosphorylées sur des résidus tyrosine lorsque les cellules sont exposées au cisaillement [19]. c-Src, une tyrosine kinase associée à la membrane dont l’activité est également réglée par la contrainte de cisaillement [4], pourrait jouer un rôle dans le processus d’activation de la FAK, favorisant l’association de FAK et de Grb2 (Figure 4).
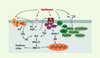 |
Figure 4. Voies de transduction mécanique menant à l’activation des MAP- kinases (mitogen activated protein kinases) ERK 1/2 (extracellular signal-regulated protein kinase 1/2) et JNK (c-jun N-terminal kinase) dans les cellules endothéliales. La contrainte de cisaillement peut stimuler soit directement, soit indirectement (via la synthèse de facteurs vasoactifs), plusieurs voies menant à l’activation des MAP-kinases. Agissant sur des récepteurs encore peu connus (?), la contrainte de cisaillement stimule la libération d’angiotensine II (AII) et de VEGF (vascular endothelial growth factor), qui interagissent ensuite avec leur récepteur respectif (couplé à une protéine G hétérotrimérique [disques orange α, β, γ] dans les cas de l’AII, à activité tyrosine kinase [RTyrK] dans le cas du VEGF). La contrainte de cisaillement peut aussi induire la phosphorylation des c-Src kinases (Src), ou stimuler les intégrines (hétérodimères αβ) qui sont fonctionnellement liées aux protéines du cytosquelette y compris FAK (focal adhesion kinase). Des phosphorylations successives mènent à l’activation de ERK 1/2 ou de JNK. PLC : phospholipase C ; IP3 : inositol 1, 4, 5-triphosphate ; DAG : diacylglycérol ; PKC : protéine kinase C ; Ras : protéine Ras ; Raf : protéine kinase Raf ; MEK1/2 : mitogen activated protein kinase kinase 1/2 ; Shc : protéine adaptatrice Shc ; Sos : son of sevenless ; Grb2 : growth factor receptor-bound protein 2 ; Cas : Crk-associated substrate. |
Quoique non encore démontrée dans le contexte de la mécanotransduction, l’activation de FAK pourrait également faire intervenir RhoA, puisque l’inhibition de cette petite protéine G désassemble les adhérences focales et réduit la phosphorylation de FAK dans des cellules endothéliales [20]. De plus, des dominants négatifs de RhoA et de la Rho kinase empêchent complètement l’alignement des cellules endothéliales soumises à un flux laminaire [21].
Cascade des MAP-kinases
La cascade des MAP-kinases (mitogen activated protein kinases) comporte plusieurs voies distinctes : ERK 1/2 (extracellular signal-regulated protein kinases 1/2), JNK (c-jun N-terminal kinase), kinase p38, et BMK1 (big mitogen-activated protein kinase 1) qui sont sensibles aux contraintes de cisaillement et qui déclenchent des réponses cellulaires différentes [22]. La phosphorylation des MAP-kinases aboutit à la régulation de l’activité de protéines cibles dans le cytoplasme et de facteurs de transcription dans le noyau.
L’activation des MAP-kinases par les contraintes mécaniques peut faire intervenir en amont les intégrines, puisque l’utilisation d’un anticorps anti-αvβ3 ou l’expression d’un dominant négatif de FAK atténuent l’activation de ERK2 et de JNK dans des cellules endothéliales stimulés par le cisaillement [23]. Toutefois, il est possible d’activer ERK à l’aide d’un mutant de l’intégrine β1, dépourvu d’un segment de la queue cytoplasmique, sans passer par l’intermédiaire de FAK [24]. La stimulation de la cascade des MAP-kinases par la contrainte de cisaillement peut également dépendre de l’activation de la protéine Gαi3 ou Gβ/γ [25], et de la phosphorylation mécanosensible des récepteurs de type tyrosine kinase [12, 25]. L’activation de Ras par les radicaux libres, qui précède en principe l’activation de la cascade de ERK1/2, a aussi été rapportée [26]. Finalement, l’inhibition de la petite protéine G RhoA ou de RhoA kinase enraye complètement l’activation de JNK par la contrainte de cisaillement [21]. Il peut donc y avoir dans un vaisseau plusieurs voies de signalisation distinctes faisant la liaison entre la stimulation mécanique et l’activation de la voie des MAP-kinases (Figure 4).
Facteurs mécaniques et profil d’expression des gènes dans les cellules vasculaires
La contrainte de cisaillement augmente l’expression des proto-oncogènes c-fos et c-jun, qui sous forme de protéines hétérodimériques AP-1 (activator protein-1), se fixent aux sites de transcription de l’ADN et agissent comme activateurs ou répresseurs transcriptionnels. De plus, SSRE (shear stress responsive element), un élément de réponse au cisaillement, a été décrit au niveau du promoteur de nombreux gènes, dont PDGF-β (platelet derived growth factor β), t-PA (tissue platelet activator), TGF-β1 (transforming growth factor β1), c-fos et c-jun [27]. D’autres facteurs de transcription des cellules endothéliales, tels que Egr-1 (early growth response-1) et Sp-1, sont également sensibles au cisaillement.
Grâce aux nouvelles techniques de génomique, l’expression différentielle des gènes sous l’effet des contraintes de cisaillement a pu être démontrée à grande échelle. Une publication récente révèle la régulation biomécanique de 52 gènes dans des cellules endothéliales de veine ombilicale en culture (HUVEC) : le cytochrome p450 et le transporteur prostaglandine sont parmi les plus puissamment stimulés, alors que le CTGF (connective tissue growth factor), l’endothéline-1 et le MCP (monocyte chemotactic protein) sont parmi les plus fortement inhibés [28]. Dans les HUVEC, M. Bongrazio et al. ont démontré l’activation de 20 gènes (dont la métalloprotéase METH-1) et l’inhibition de 13 gènes, dont l’angiopoïétine 2 et le GAS3 (growth-arrest specific gene 3), par la contrainte de cisaillement [29]. Une troisième étude décrit la surexpression de la métalloprotéase-1, et des récepteurs de l’angiopoïétine (Tie-2) et du VEGF (FLK-1) dans des cellules endothéliales aortiques humaines soumises à un flux laminaire, et la réduction simultanée de l’expression de gènes codant pour des protéines impliquées dans l’inflammation et la prolifération [30]. Enfin, dans des HUVEC, 143 gènes dont l’expression variait en fonction des conditions de culture imposées, comme l’absence de contraintes de cisaillement, résultant de l’écoulement laminaire ou turbulent, ont été classés selon leur rôle connu dans la mécanotransduction, la réponse à l’agression ou l’athérogenèse [31].
Toutefois, dans la mesure où l’environnement matriciel et le phénotype cellulaire sont des paramètres déterminants de la réponse des cellules vasculaires à la stimulation mécanique, le profil d’expression des gènes obtenu sur cellules endothéliales en culture ne reflète certainement pas de façon fidèle ce qui se passe in vivo. Une étude a même démontré l’importance de distinguer les préparations artérielles des préparations veineuses, puisque l’expression des gènes est très différente d’un type cellulaire à l’autre [32]. Quelques travaux récents ont fait appel à des modèles de vaisseaux en culture ou in vivo pour élucider l’expression différentielle de gènes sous différentes conditions de flux. Un flux élevé dans des artères in vivo induit la surexpression soutenue de PDGF-A et PDGF-B dans les cellules endothéliales [33]. Par ailleurs, la contrainte de cisaillement induit l’expression de furine et de TGFβ dans l’endothélium de la carotide de lapin à haut débit [34]. Dans des veines ombilicales perfusées, c-jun est stimulé par un cisaillement élevé [35], de même que la prostacycline synthase, la thromboxane synthase, les cyclo-oxygénases COX-1 et -2 [36], à l’inverse de l’expression du VEGF qui chute dans les mêmes conditions [37].
Conclusions
Les vaisseaux sanguins possèdent des mécanismes qui leur permettent de réagir immédiatement à des variations locales des contraintes de cisaillement (qui augmentent avec le débit sanguin). En revanche, si ces mécanismes sont insuffisants pour compenser les altérations de flux, le phénotype des cellules vasculaires se modifie et entraîne des changements locaux de trophicité ramenant, à plus long terme, les contraintes mécaniques à des valeurs physiologiques. Ce remodelage vasculaire fait intervenir au niveau des cellules endothéliales de nombreuses protéines qui sont impliquées dans la perception de la contrainte mécanique et la transmission du signal dans la cellule, ce qui conduit à l’expression différentielle de gènes contrôlant la structure ou la fonction des vaisseaux.
Dans le cas d’un écoulement laminaire, la contrainte de cisaillement s’exprime comme :tau= 4µQ/π r3 où µ est la viscosité, Q le débit et r le rayon du vaisseau.
Références
- Glagov S. Intimal hyperplasia, vascular remodeling, and restenosis problem. Circulation 1994; 89 : 2888–91. [Google Scholar]
- Tronc F, Wassef M, Esposito B, et al. Role of NO in flow-induced remodeling of the rabbit common carotid artery. Arterioscler Thromb Vasc Biol 1996; 16 : 1256–62. [Google Scholar]
- Tronc F, Mallat Z, Lehoux S, et al. Role of matrix metalloproteinases in blood flow-induced arterial enlargement. Arterioscler Thromb Vasc Biol 2000; 20 : E120–6. [Google Scholar]
- Jalali S, del Pozo MA, Chen K, et al. Integrin-mediated mechanotransduction requires its dynamic interaction with specific extracellular matrix (ECM) ligands. Proc Natl Acad Sci USA 2001; 98 : 1042–6. [Google Scholar]
- Muller JM, Chilian WM, Davis MJ. Integrin signaling transduces shear stress-dependent vasodilation of coronary arterioles. Circ Res 1997; 80 : 320–6. [Google Scholar]
- Bhullar IS, Li YS, Miao H, et al. Fluid shear stress activation of IkappaB kinase is integrin-dependent. J Biol Chem 1998; 273 : 30544–9. [Google Scholar]
- Hoger JH, Ilyin VI, Forsyth S, Hoger A. Shear stress regulates the endothelial Kir2.1 ion channel. Proc Natl Acad Sci USA 2002; 99 : 7780–5. [Google Scholar]
- Ohno M, Gibbons GH, Dzau VJ, Cooke JP. Shear stress elevated endothelial cGMP. Role of a potassium channel and G protein coupling. Circulation 1993; 88 : 193–7. [Google Scholar]
- Gudi SR, Clark CB, Frangos JA. Fluid flow rapidly activates G proteins in human endothelial cells. Involvement of G proteins in mechanochemical signal transduction. Circ Res 1996; 79 : 834–9. [Google Scholar]
- Hsieh HJ, Li NQ, Frangos JA. Pulsatile and steady flow induces c-fos expression in human endothelial cells. J Cell Physiol 1993; 154 : 143–51. [Google Scholar]
- Hansen CA, Schroering AG, Carey DJ, Robishaw JD. Localization of a heterotrimeric G protein gamma subunit to focal adhesions and associated stress fibers. J Cell Biol 1994; 126 : 811–29. [Google Scholar]
- Chen KD, Li YS, Kim M, et al. Mechanotransduction in response to shear stress. Roles of receptor tyrosine kinases, integrins, and Shc. J Biol Chem 1999; 274 : 18393–400. [Google Scholar]
- De Keulenaer GW, Chappell DC, Ishizaka N, et al. Oscillatory and steady laminar shear stress differentially affect human endothelial redox state : role of a superoxide-producing NADH oxidase. Circ Res 1998; 82 : 1094–101. [Google Scholar]
- Silacci P, Desgeorges A, Mazzolai L, et al. Flow pulsatility is a critical determinant of oxidative stress in endothelial cells. Hypertension 2001; 38 : 1162–6. [Google Scholar]
- Dimmeler S, Fleming I, Fisslthaler B, et al. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 1999; 399 : 601–5. [Google Scholar]
- Dimmeler S, Assmus B, Hermann C, et al. Fluid shear stress stimulates phosphorylation of Akt in human endothelial cells : involvement in suppression of apoptosis. Circ Res 1998; 83 : 334–41. [Google Scholar]
- Govers R, Rabelink TJ. Cellular regulation of endothelial nitric oxide synthase. Am J Physiol Renal Physiol 2001; 280 : F193–206. [Google Scholar]
- Mallat Z, Tedgui A. Apoptosis in the vasculature : Mechanisms and functional importance. Br J Pharmacol 2000; 130 : 947–62. [Google Scholar]
- Ishida T, Peterson TE, Kovach NL, Berk BC. MAP kinase activation by flow in endothelial cells : role of beta 1 integrins and tyrosine kinases. Circ Res 1996; 79 : 310–6. [Google Scholar]
- Carbajal JM, Schaeffer RC, Jr. RhoA inactivation enhances endothelial barrier function. Am J Physiol 1999; 277 : C955–64. [Google Scholar]
- Li S, Chen BP, Azuma N, et al. Distinct roles for the small GTPases Cdc42 and Rho in endothelial responses to shear stress. J Clin Invest 1999; 103 : 1141–50. [Google Scholar]
- Lehoux S, Tedgui A. Signal transduction of mechanical stresses in the vascular wall. Hypertension 1998; 32 : 338–45. [Google Scholar]
- Li S, Kim M, Hu YL, et al. Fluid shear stress activation of focal adhesion kinase. Linking to mitogen-activated protein kinases. J Biol Chem 1997; 272 : 30455–62. [Google Scholar]
- Barberis L, Wary KK, Fiucci G, et al. Distinct roles of the adaptor protein Shc and focal adhesion kinase in integrin signaling to ERK. J Biol Chem 2000; 275 : 36532–40. [Google Scholar]
- Jo H, Sipos K, Go YM, et al. Differential effect of shear stress on extracellular signal-regulated kinase and N-terminal Jun kinase in endothelial cells. Gi2- and Gbeta/gamma-dependent signaling pathways. J Biol Chem 1997; 272 : 1395–401. [Google Scholar]
- Abe J, Okuda M, Huang Q, et al. Reactive oxygen species activate p90 ribosomal S6 kinase via Fyn and Ras. J Biol Chem 2000; 275 : 1739–48. [Google Scholar]
- Resnick N, Collins T, Atkinson W, et al. Platelet-derived growth factor B chain promoter contains a cis-acting fluid shear-stress-responsive-element. Proc Natl Acad Sci USA 1993; 90 : 4591–5. [Google Scholar]
- McCormick SM, Eskin SG, McIntire LV, et al. DNA microarray reveals changes in gene expression of shear stressed human umbilical vein endothelial cells. Proc Natl Acad Sci USA 2001; 98 : 8955–60. [Google Scholar]
- Bongrazio M, Baumann C, Zakrzewicz A, et al. Evidence for modulation of genes involved in vascular adaptation by prolonged exposure of endothelial cells to shear stress. Cardiovasc Res 2000; 47 : 384–93. [Google Scholar]
- Chen BP, Li YS, Zhao Y, et al. DNA microarray analysis of gene expression in endothelial cells in response to 24-h shear stress. Physiol Genomics 2001; 7 : 55–63. [Google Scholar]
- Garcia-Cardena G, Comander J, Anderson KR, et al. Biomechanical activation of vascular endothelium as a determinant of its functional phenotype. Proc Natl Acad Sci USA 2001; 98 : 4478–85. [Google Scholar]
- Adams LD, Geary RL, McManus B, Schwartz SM. A comparison of aorta and vena cava medial message expression by cDNA array analysis identifies a set of 68 consistently differentially expressed genes, all in aortic media. Circ Res 2000; 87 : 623–31. [Google Scholar]
- Tulis DA, Prewitt RL. Medial and endothelial platelet-derived growth factor A chain expression is regulated by in vivo exposure to elevated flow. J Vasc Res 1998; 35 : 413–20. [Google Scholar]
- Negishi M, Lu D, Zhang YQ, et al. Upregulatory expression of furin and transforming growth factor-beta by fluid shear stress in vascular endothelial cells. Arterioscler Thromb Vasc Biol 2001; 21 : 785–90. [Google Scholar]
- Gan L, Doroudi R, Hagg U, et al. Differential immediate-early gene responses to shear stress and intraluminal pressure in intact human conduit vessels. FEBS Lett 2000; 477 : 89–94. [Google Scholar]
- Doroudi R, Gan LM, Selin Sjogren L, Jern S. Effects of shear stress on eicosanoid gene expression and metabolite production in vascular endothelium as studied in a novel biomechanical perfusion model. Biochem Biophys Res Commun 2000; 269 : 257–64. [Google Scholar]
- Gan L, Miocic M, Doroudi R, et al. Distinct regulation of vascular endothelial growth factor in intact human conduit vessels exposed to laminar fluid shear stress and pressure. Biochem Biophys Res Commun 2000; 272 : 490–6. [Google Scholar]
Liste des figures
|
Figure 1. Étapes conduisant au remodelage vasculaire induit par la contrainte de cisaillement. La contrainte de cisaillement induit la production de radicaux libres oxygénés (O2‾), la libération de monoxyde d’azote (NO) et la synthèse de métalloprotéases matricielles (MMP) par les cellules endothéliales. L’interaction de NO avec O2‾ produit du peroxynitrite, qui active les MMP. Ces dernières dégradent la matrice extracellulaire, provoquant d’une part l’apoptose des cellules ayant perdu leurs attaches matricielles, et d’autre part l’augmentation du calibre des vaisseaux. Le NO peut aussi, avec le concours des facteurs de croissance stimulés par la contrainte de cisaillement, participer à l’hyperplasie des cellules vasculaires. CML : cellules musculaires lisses. |
| Dans le texte | |
|
Figure 2. Mécanosenseurs dans les cellules endothéliales. Les cellules endothéliales possèdent à leur surface plusieurs récepteurs susceptibles d’être activés par la contrainte de cisaillement, tels que les récepteurs à activité tyrosine kinase (RTyrK), les intégrines (hétérodimères α et β), les canaux potassiques, les protéines G hétérotrimériques (αβγ), et la NAD(P)H oxydase. L’activation d’un ou de plusieurs de ces récepteurs peut conduire à l’entrée de calcium ou à l’activation de différentes cascades biochimiques. Ainsi, l’activation de la NO synthase par la contrainte de cisaillement (et la libération secondaire de monoxyde d’azote [NO]) nécessite une phosphorylation par la protéine kinase Akt (Akt). Shc : protéine adaptatrice Shc ; MAPK : voie des MAP-kinases (mitogen activated protein kinases). |
| Dans le texte | |
|
Figure 3. Monoxyde d’azote (NO), protéine kinase Akt (Akt) et apoptose. L’activation de la protéine kinase Akt protège la cellule endothéliale de l’apoptose en inhibant la voie de caspases, soit en activant la synthèse de NO par l’intermédiaire de la NO synthase (eNOS), soit en phosphorylant Bad (Bcl-2-antagonist of cell death), empêchant sa liaison avec Bcl-2 (B-cell CLL/lymphoma 2) ou Bcl-XL (Bcl-2-like 1) à la membrane des mitochondries. L’activation d’AKT peut également activer l’expression de gènes favorisant la survie cellulaire. Flk-1 : récepteur à activité tyrosine kinase Flk-1 ; c-Src : kinases c-Src ; PI3K : phosphatidylinositol-3-kinase. |
| Dans le texte | |
|
Figure 4. Voies de transduction mécanique menant à l’activation des MAP- kinases (mitogen activated protein kinases) ERK 1/2 (extracellular signal-regulated protein kinase 1/2) et JNK (c-jun N-terminal kinase) dans les cellules endothéliales. La contrainte de cisaillement peut stimuler soit directement, soit indirectement (via la synthèse de facteurs vasoactifs), plusieurs voies menant à l’activation des MAP-kinases. Agissant sur des récepteurs encore peu connus (?), la contrainte de cisaillement stimule la libération d’angiotensine II (AII) et de VEGF (vascular endothelial growth factor), qui interagissent ensuite avec leur récepteur respectif (couplé à une protéine G hétérotrimérique [disques orange α, β, γ] dans les cas de l’AII, à activité tyrosine kinase [RTyrK] dans le cas du VEGF). La contrainte de cisaillement peut aussi induire la phosphorylation des c-Src kinases (Src), ou stimuler les intégrines (hétérodimères αβ) qui sont fonctionnellement liées aux protéines du cytosquelette y compris FAK (focal adhesion kinase). Des phosphorylations successives mènent à l’activation de ERK 1/2 ou de JNK. PLC : phospholipase C ; IP3 : inositol 1, 4, 5-triphosphate ; DAG : diacylglycérol ; PKC : protéine kinase C ; Ras : protéine Ras ; Raf : protéine kinase Raf ; MEK1/2 : mitogen activated protein kinase kinase 1/2 ; Shc : protéine adaptatrice Shc ; Sos : son of sevenless ; Grb2 : growth factor receptor-bound protein 2 ; Cas : Crk-associated substrate. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.