")
")
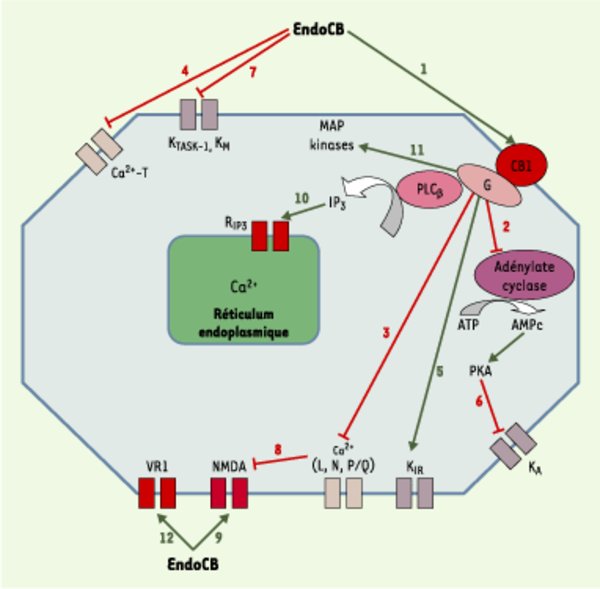
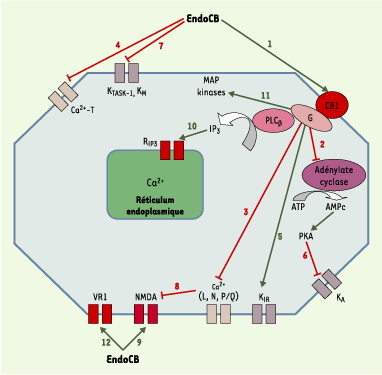
Figure 2.
Télécharger l'image originale
{kind=link}
Principales voies de transduction du signal modifiées après action des endocannabinoïdes (endoCB) via les récepteurs CB1, VR1 ou par action directe. L’activation des récepteurs CB1 et CB2 par les endoCB (1) conduit à une inhibition de l’activité cyclasique (2). Cet effet est inhibé par un traitement par la toxine pertussique, révélant ainsi l’intervention d’une protéine G de type Gi/o dans le couplage récepteur-enzyme [14, 15]. L’anandamide (AEA) et le 2-arachidonoyl glycérol (2-AG) sont des agonistes entiers de l’inhibition de l’adénylate cyclase via le CB1. En revanche, seul le 2-AG est agoniste entier au niveau du CB2. Dans certaines conditions, l’activation du récepteur CB1 (et pas le CB2) conduit à une production d’AMPc via une protéine de type Gs. Dans ce cas, l’AEA devient un agoniste partiel du CB1, contrairement au 2-AG qui reste agoniste entier. L’activation du récepteur CB1 provoque également une inhibition indirecte, par le biais d’une protéine Gi/o mais indépendante de l’activité cyclasique, des canaux Ca2+ sensibles au potentiel de type N, L et Q/P [14, 15] (3), et une inhibition directe des canaux de type T [16] (4). Les endoCB augmentent par ailleurs l’activité des canaux potassiques de la rectification entrante (KIR) (5), par le biais d’une protéine G de type Gi/o, mais indépendamment de l’inhibition de l’adénylate cyclase. L’activation des récepteurs CB1 diminue la sensibilité au potentiel de membrane des canaux potassiques de type A (KA) (6), via une protéine Gi/o et de façon dépendante de l’inhibition de la voie adénylate cyclase/protéine kinase A (PKA) [14, 15]. De plus, les endoCB inhibent deux autres types de canaux potassiques : des canaux de fuite sensibles aux protons (TASK-1) et les canaux de type M (KM) [17, 18] (7). L’AEA a un double effet, inhibiteur et stimulateur, sur les récepteurs du glutamate de type NMDA (M-méthyl-D-aspartate) [14, 15]. L’inhibition est une conséquence indirecte de l’inhibition des conductances Ca2+ de type P/Q après activation du CB1 (8), tandis que l’activation résulte d’un effet direct des endoCB sur le récepteur NMDA (9), conduisant à une augmentation de l’influx calcique à travers le canal. L’AEA inhibe la perméabilité des jonctions communicantes entre les astrocytes, via une protéine G de type Gi/o couplée à un récepteur de type CB différent de CB1 ; il a pour conséquence une inhibition du couplage électrique et de la propagation des vagues calciques intercellulaires [19]. Par ailleurs, les endoCB, après activation du récepteur CB1 et activation subséquente de la phospholipase Cβ (PLCβ), stimulent la mobilisation du Ca2+ intracellulaire stocké dans le réticulum endoplasmique des neurones [14, 15] et des astrocytes (10) [20]. L’activation par les endoCB de la voie des MAP kinases (mitogen-activated protein kinases) [14, 15] (11) déclenche une cascade aboutissant in fine à l’activation de facteurs de transcription multiples, tels que krox 24, c-fos ou c-jun. Deux autres MAP kinases sont activées par les endoCB lors de stress cellulaires : p38-MAPK et c-jun-N-terminal kinase (JNK). L’activation de JNK conduit à une apoptose cellulaire, alors que celle de p38-MAPK a des effets neuroprotecteurs, ainsi qu’anti-profilérateurs au niveau de cellules tumorales [21, 22]. L’action des endoCB sur la voie des MAP kinases constitue un champ de recherche extrêmement prometteur au niveau thérapeutique (voir plus loin), car cette voie intervient dans le devenir de la cellule (processus de différenciation morphologique et de survie neuronale). Seule l’AEA, parmi les endoCB, active le récepteur vanilloïde de type 1 (VR1) (12), un canal cationique non sélectif de la famille des canaux TRP (transient receptor potential) impliqués dans les phénomènes de détection de stimulus nocicepteurs et dans la transduction de l’hyperalgésie inflammatoire et thermique. Un agoniste de VR1, l’olvanil (voir Figure 1), agit comme un agoniste partiel du CB1, ce qui suggère l’existence d’un recouvrement partiel, dans la reconnaissance des ligands, entre les récepteurs VR1 et CB1. Les affinités de couplage des récepteurs CB1 et CB2 aux protéines G ne sont pas équivalentes : si les deux types de récepteurs possèdent une très forte affinité pour Gi, celle du CB1 pour Go est 10 fois supérieure à celle du CB2. Cela peut expliquer l’absence, ou la faiblesse, d’interaction du CB2 avec les canaux ioniques, contrairement au CB1 qui est capable de moduler l’activité de nombreux canaux. Les endoCB inhibent, directement ou indirectement (via le CB1), la perméabilité de canaux calciques préférentiellement présynaptiques (comme les récepteurs CB1) et impliqués dans le contrôle de la libération des neurotransmetteurs.
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.