")
")
| Issue |
Med Sci (Paris)
Volume 19, Number 10, Octobre 2003
|
|
|---|---|---|
| Page(s) | 999 - 1002 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/20031910999 | |
| Published online | 15 octobre 2003 | |
Bases cellulaires des transitions de l’état de sommeil aux paroxysmes épileptiformes
Cellular basis of transition between sleep and electrographic seizures
Laboratoire de neurophysiologie, Département d’anatomie et physiologie, Faculté de médecine, Université Laval, Québec, G1K 7P4 Canada
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Chez l’homme, certaines crises épileptiques apparaissent pendant le sommeil à ondes lentes. Nos recherches expérimentales ont démontré une transformation de l’activité cérébrale électrique qui caractérise le sommeil vers des paroxysmes de type épileptique. L’origine des crises se situe dans le cortex cérébral. Les paroxysmes se propagent d’un groupe de neurones à un autre, avant de déboucher dans le thalamus. Les neurones thalamiques de relais sont inhibés pendant les crises corticales, ce qui pourrait expliquer la perte de conscience dans l’épilepsie de type absence (petit-mal).
Abstract
Epileptic seizures mainly develop during slow-wave sleep. Our experiments, using multi-site, extra- and intracellular recordings, show a transformation without discontinuity from sleep patterns to seizures. The cerebral cortex is the minimal substrate of paroxysms with spike-wave complexes at ~3 Hz. Simultaneously, thalamocortical neurons are steadily inhibited and cannot relay signals from the outside world to cortex. This may explain the unconsciousness during certain types of epilepsy.
© 2003 médecine/sciences - Inserm / SRMS
Le sommeil peut être divisé en deux phases principales, le sommeil à ondes lentes et le sommeil avec mouvements oculaires rapides, ou sommeil paradoxal. Chez l’homme, différents types de crises épileptiques, comme les absences (petit-mal) ou le syndrome Lennox- Gastaut, apparaissent préférentiellement pendant le sommeil à ondes lentes [1, 2]. Cette relation entre un état normal caractérisé par la baisse du niveau de vigilance et un phénomène pathologique a également été établie dans certains modèles animaux; par exemple, pendant les périodes de somnolence ou de sommeil léger, des macaques en expérimentation chronique montrent des crises épileptiques du type pointe-onde à ~3 Hz, accompagnées de signes oculaires, ressemblant au tableau clinique de l’épilepsie petit-mal [3]. Dans la même étude, l’apparition des pointes-ondes et des décharges neuronales dans la profondeur du cortex, sans reflet à la surface corticale, avait suggéré une origine corticale pour ces crises [3]. Cette hypothèse contraste avec l’idée conventionnelle qui suggère une genèse de l’épilepsie petit-mal au niveau d’un système situé en profondeur dans le cerveau, censé produire des décharges épileptiques qui, suivant une expression classique, seraient synchronisées bilatéralement d’une manière soudaine [4]. L’existence d’un tel système, ayant les propriétés d’un pacemaker avec des projections bilatérales qui pourraient expliquer l’apparition simultanée des décharges épileptiques dans les deux hémisphères, n’a jamais été confirmée. En réalité, la simultanéité des décharges épileptiques dès le début de la crise n’est visible que macroscopiquement, à l’examen de l’électroencéphalogramme (EEG) qui ne peut pas révéler les délais des jonctions interneuronales. L’analyse des activités cellulaires met en évidence une progression qui débute dans certains groupes neuronaux du néocortex pour envahir différents territoires du cerveau seulement après un certain délai.
Nous avons abordé ce problème chez des chats anesthésiés ou en expérience chronique, en utilisant des enregistrements multiples, y compris des enregistrements intracellulaires simultanés de paires de neurones corticaux ou corticaux et thalamiques. Nos études montrent que le substrat minimum des crises électrographiques avec complexes pointe-onde à ~3 Hz est le cortex cérébral, car les paroxysmes apparaissent dans le cortex des chats ayant subi une lésion extensive du thalamus [5]. Des crises du type pointe-onde ou du type Lennox-Gastaut (qui comportent aussi des épisodes d’ondes plus rapides, à 10-20 Hz) (Figure 1) se propagent à travers des circuits corticaux mono-, oligo- ou multisynaptiques, avec des latences entre différents groupes neuronaux allant de 3-10 ms à 50- 100 ms [6]. Le thalamus n’est envahi que beaucoup plus tard, après quelques secondes [7].
 |
Figure 1. Transformation de l’activité électrique du sommeil à ondes lentes vers une activité paroxystique de type crise Lennox-Gastaut. Enregistrement intracellulaire d’un neurone de type pyramidal dans l’aire corticale 5 (A) et de l’EEG (électroencéphalogramme) dans la profondeur de l’aire 5 (B). Les différentes époques illustrées montrent l’oscillation lente du sommeil, se développant progressivement vers une crise du type pointe-onde interrompue par une brève période d’ondes rapides, après laquelle la crise se termine. De courtes époques des trois périodes sont représentées plus bas, d’une façon plus détaillée (modifié d’après [8]). |
Ces données réfutent l’hypothèse d’une origine thalamique (ou centrencéphalique) de l’épilepsie petit-mal. Elles indiquent que l’avalanche en série des ensembles cellulaires corticaux implique les jonctions interneuronales. La progression de l’activité EEG témoignant du sommeil à ondes lentes vers la crise d’épilepsie est progressive et la synchronie normale du sommeil se transforme, sans discontinuité, en hypersynchronie paroxystique. La transformation progressive des patrons de sommeil vers la crise épileptique est corroborée par une grande similitude entre les relations temporelles des potentiels de champ (EEG) et intracellulaires pendant le sommeil et pendant la crise (Figure 1) [8].
Étant donné que les cellules gliales ne sont plus considérées seulement comme des éléments qui reflètent passivement l’activité neuronale, mais qui possède des récepteurs pour différents neurotransmetteurs et des propriétés qui peuvent leur conférer un rôle actif dans l’activité électrique du cerveau [9], un dialogue entre les neurones et les cellules gliales dans la genèse des activités épileptiques pendant le sommeil lent a pu aussi être envisagé. Cela a été démontré par des enregistrements intracellulaires simultanés de ces deux types de cellules, et il a été suggéré que les cellules gliales contribuent au décours temporel des événements neuronaux paroxystiques [10, 11].
Les deux composantes des phénomènes paroxystiques, la pointe et l’onde, sont produites par des mécanismes différents. La pointe de l’EEG est associée à des décharges de neurones pyramidaux excitateurs ainsi qu’à celles de neurones locaux inhibiteurs [12]. L’idée d’une participation simultanée des neurones inhibiteurs modifie le concept selon lequel la pointe n’est qu’une sommation géante de potentiels excitateurs [13]. Par ailleurs, l’onde EEG, pendant laquelle les neurones corticaux sont silencieux, n’est pas due à une inhibition active, mais à une disfacilitation des réseaux corticaux (enlèvement des afférences, principalemnent celles des systèmes excitateurs) et à certains courants potassiques. Le passage du silence neuronal pendant l’onde EEG vers l’explosion épileptique qui caractérise la pointe EEG est dû à un courant ionique qui est activé par l’hyperpolarisation du neurone pendant l’onde et qui dépolarise le neurone pour aboutir finalement à la pointe épileptique [14].
Pendant l’orage épileptique d’origine corticale, le comportement des deux types majeurs de neurones thalamiques est dissemblable. Ces deux types cellulaires sont les neurones excitateurs thalamocorticaux ou neurones de relais, qui transfèrent vers le cortex les signaux du monde extérieur, et les neurones thalamiques réticulaires qui ne projettent pas au cortex, utilisent l’acide γ-aminobutyrique (GABA) comme transmetteur et constituent une boucle de rétroaction inhibitrice avec les neurones de relais. Ces connexions ainsi que les rapports des neurones corticaux avec les neurones thalamiques sont illustrés dans la Figure 2. Chaque décharge synchrone des neurones corticaux pendant la pointe épileptique excite les neurones réticulaires inhibiteurs, qui répondent avec des bouffées de potentiels d’action prolongées [15, 16]. En conséquence, les neurones de relais, qui sont les cibles des neurones réticulaires, sont inhibés d’une manière soutenue (Figure 2). Ces données, qui proviennent de nos expériences sur des modèles d’absence et de syndrome Lennox-Gastaut, ont été corroborées par des enregistrements thalamiques chez des rats avec un modèle génétique d’absence épileptique [17, 18]. L’inhibition des neurones thalamiques de relais pendant les crises électriques avec des complexes pointe-onde pourrait être à la base de la perte de conscience et des relations avec le monde extérieur pendant les crises d’absence ou petit-mal.
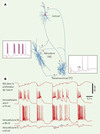 |
Figure 2. Relations des neurones corticaux et thalamiques pendant une crise de type pointeonde. A. Trois neurones (corticothalamique, thalamique réticulaire-RE, et thalamocortical-TC) ont été enregistrés et colorés intracellulairement. Les signes de leur fonction (excitatrice ou inhibitrice) sont indiqués par des «plus» ou des «moins». Les parties encadrées représentent les réponses des neurones RE et TC à une stimulation corticale (excitation du neurone RE, inhibition du neurone TC). B. Pendant une crise pointe-onde corticale (visible aussi bien sur l’EEG (électroencéphalogramme) qu’au niveau de l’enregistrement intracellulaire), le neurone inhibiteur RE est excité, tandis que le neurone cible TC est inhibé (les deux cercles bleus représentent des potentiels inhibiteurs postsynaptiques) (modifié d’après [23]). |
Les données présentées ici peuvent poser la question de savoir à quel niveau cérébral agit l’une des substances pharmacologiques les plus puissantes dans le traitement clinique de l’épilepsie petit-mal, l’éthosuximide. Des travaux antérieurs ont suggéré que cette substance bloque un courant calcique à seuil bas qui est évident au niveau des neurones thalamiques [19], ce qui avait renforcé l’hypothèse d’une origine thalamique des absences épileptiques. On sait maintenant que ce courant calcique à seuil bas est également exprimé par les neurones corticaux, pyramidaux aussi bien que les interneurones locaux inhibiteurs [20, 21]. À la lumière de nos observations expérimentales, démontrant la genèse corticale des crises pointe-onde, nous avons suggéré que l’éthosuximide agit au niveau des neurones corticaux [5].
Les relations complexes entre le cortex et le thalamus, avec excitation corticale directe des neurones réticulaires et inhibition indirecte des neurones thalamiques de relais, à travers l’excitation initiale des neurones réticulaires, plaident pour l’idée [22] que des recherches sur des phénomènes aussi globaux que le sommeil et l’épilepsie doivent s’effectuer chez des animaux au cerveau intact, avec des connexions corticothalamiques fonctionnelles, sous le contrôle des systèmes modulateurs du tronc cérébral et du prosencéphale basal.
Références
- Niedermeyer E. Epileptic seizure disorders. In: Niedermeyer E, Lopes da Silva F, eds. Electroencephalography: basic principles, clinical applications and related fields. Baltimore: Williams and Wilkins, 1999: 476–585. [Google Scholar]
- Steriade M. Neuronal substrates of sleep and epilepsy. Cambridge (United Kingdom): Cambridge University Press, 2003: 522 p. [Google Scholar]
- Steriade M. Interneuronal epileptic discharges related to spike-and-wave cortical seizures in behaving monkeys. Electroencephalogr Clin Neurophysiol 1974; 37: 247–63. [Google Scholar]
- IFSECN. A glossary of terms most commonly used by clinical electroencephalographers. Electroencephalogr Clin Neurophysiol 1974; 37: 538–48. [Google Scholar]
- Steriade M, Contreras D. Spike-wave complexes and fast runs of cortically generated seizures. I. Role of neocortex and thalamus. J Neurophysiol 1998; 80: 1439–55. [Google Scholar]
- Steriade M, Amzica F. Dynamic coupling among neocortical neurons during evoked and spontaneous spike-wave seizure activity. J Neurophysiol 1994; 72: 2051–69. [Google Scholar]
- Neckelmann D, Amzica F, Steriade M. Spike-wave complexes and fast components of cortically generated seizures. III. Synchronizing mechanisms. J Neurophysiol 1998; 80: 1480–94. [Google Scholar]
- Steriade M, Amzica F, Neckelmann D, Timofeev I. Spike-wave complexes and fast runs of cortically generated seizures. II. Extra- and intracellular patterns. J Neurophysiol 1998; 80: 1456–79. [Google Scholar]
- Kettenmann H, Ransom BR. Neuroglia. New York: Oxford University Press, 1995: 1079. [Google Scholar]
- Amzica F, Steriade M. Neuronal and glial membrane potentials during sleep and paroxysmal oscillations in the cortex. J Neurosci 2000; 20: 6648–65. [Google Scholar]
- Amzica F, Massimini M, Manfridi A. Spatial buffering during slow and paroxysmal oscillations in cortical networks of glial cells in vivo. J Neurosci 2002; 22: 1042–53. [Google Scholar]
- Timofeev I, Grenier F, Steriade M. The role of chloride-dependent inhibition and the activity of fast-spiking neurons during cortical spike-wave seizures. Neuroscience 2002; 114: 1115–32. [Google Scholar]
- Johnston D, Brown TH. Giant spike potential hypothesis for epileptiform activity. Science 1981; 211: 294–7. [Google Scholar]
- Timofeev I, Bazhenov M, Sejnowski TJ, Steriade M. Cortical Ih takes part in the generation of paroxysmal activities. Proc Natl Acad Sci USA 2002; 99: 9533–7. [Google Scholar]
- Steriade M, Contreras D. Relations between cortical and thalamic cellular events during transition from sleep pattern to paroxysmal activity. J Neurosci 1995; 15: 623–42. [Google Scholar]
- Timofeev I, Grenier F, Steriade M. Spike-wave complexes and fast runs of cortically generated seizures. IV. Paroxysmal fast runs in cortical and thalamic neurons. J Neurophysiol 1998; 80: 1495–513. [Google Scholar]
- Pinault D, Leresche N, Charpier S, et al. Intracellular recordings in thalamic neurones during spontaneous spike and wave discharges in rats with absence epilepsy. J Physiol 1998; 509: 449–56. [Google Scholar]
- Crunelli V, Leresche N. Childhood absence epilepsy: genes, channels, neurons and networks. Nat Rev Neurosci 2002; 3: 371–82. [Google Scholar]
- Coulter DA, Huguenard JR, Prince DA. Characterization of ethosuximide reduction of low-threshold calcium current in thalamic neurons. Ann Neurol 1989; 25: 582–93. [Google Scholar]
- de la Peña E, Geijo- Barrientos E. Laminar localization, morphology, and physiological properties of pyramidal neurons that have lowthreshold calcium current in the guinea-pig medial frontal cortex. J Neurosci 1996; 16: 5301–11. [Google Scholar]
- Destexhe A, Contreras D, Steriade M. LTS cells in cerebral cortex and their role in generating spikeand- wave oscillations. Neurocomputing 2001; 38-40: 555–63. [Google Scholar]
- Steriade M. The intact and sliced brain. Cambridge (Massachusetts): The MIT Press, 2001: 366 p. [Google Scholar]
- Steriade M. The GABAergic reticular nucleus: a preferential target of corticothalamic projctions. Proc Natl Acad Sci USA 2001; 98: 3625–7. [Google Scholar]
Liste des figures
|
Figure 1. Transformation de l’activité électrique du sommeil à ondes lentes vers une activité paroxystique de type crise Lennox-Gastaut. Enregistrement intracellulaire d’un neurone de type pyramidal dans l’aire corticale 5 (A) et de l’EEG (électroencéphalogramme) dans la profondeur de l’aire 5 (B). Les différentes époques illustrées montrent l’oscillation lente du sommeil, se développant progressivement vers une crise du type pointe-onde interrompue par une brève période d’ondes rapides, après laquelle la crise se termine. De courtes époques des trois périodes sont représentées plus bas, d’une façon plus détaillée (modifié d’après [8]). |
| Dans le texte | |
|
Figure 2. Relations des neurones corticaux et thalamiques pendant une crise de type pointeonde. A. Trois neurones (corticothalamique, thalamique réticulaire-RE, et thalamocortical-TC) ont été enregistrés et colorés intracellulairement. Les signes de leur fonction (excitatrice ou inhibitrice) sont indiqués par des «plus» ou des «moins». Les parties encadrées représentent les réponses des neurones RE et TC à une stimulation corticale (excitation du neurone RE, inhibition du neurone TC). B. Pendant une crise pointe-onde corticale (visible aussi bien sur l’EEG (électroencéphalogramme) qu’au niveau de l’enregistrement intracellulaire), le neurone inhibiteur RE est excité, tandis que le neurone cible TC est inhibé (les deux cercles bleus représentent des potentiels inhibiteurs postsynaptiques) (modifié d’après [23]). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.