")
")
| Issue |
Med Sci (Paris)
Volume 18, Number 12, Décembre 2002
|
|
|---|---|---|
| Page(s) | 1257 - 1265 | |
| Section | M/S Revues : Articles de Synthèse | |
| DOI | https://doi.org/10.1051/medsci/200218121257 | |
| Published online | 15 décembre 2002 | |
Les statines en thérapeutique cardiovasculaire
Pleiotropic effects of statins: Implications in the treatment of cardiovascular diseases
Inserm EPI 99-36, Faculté de Médecine, 27, boulevard Jean Moulin, 13385 Marseille Cedex 5, France
*
Gilles.Nalbone @medecine.univ-mrs.fr
Résumé
Les dyslipidémies athérogènes sont responsables de la plupart des accidents cardiovasculaires ischémiques, qui restent dans les pays industrialisés une des causes majeures de morbidité et de mortalité. Une conduite nutritionnelle adaptée permet parfois de corriger l’excès de lipides circulants, mais lorsqu’elle s’avère insuffisante, la thérapeutique médicamenteuse s’impose. Les inhibiteurs de l’hydroxy-méthylglutaryl-coenzyme A (HMG-CoA) réductase, ou statines, ont il y a plus de dix ans révolutionné le traitement de l’hypercholestérolémie. Cependant, il semble de plus en plus évident que le bénéfice thérapeutique de cette classe de médicaments dépasse celui imputable à la seule baisse du cholestérol circulant puisque les statines, de par leur mode d’action intracellulaire, modulent favorablement l’expression et l’activité de différentes protéines impliquées dans la fonction vasculaire. Grâce à leur extrême diversité d’action sur le compartiment vasculaire, ces molécules sont bien sûr un atout thérapeutique essentiel, mais représentent également un outil pharmacologique pouvant aider le chercheur à évaluer les modifications post-traductionnelles des protéines de signalisation, ainsi que les conséquences de ces modifications sur la régulation de l’expression des gènes.
Abstract
Statins lower circulating cholesterol by blocking the activity of HMG-CoA reductase, the rate-limiting enzyme in cholesterol synthesis. However, statins pos-sess pleiotropic properties that complement their cholesterol lowering effect, and makes this class of drugs very efficient to prevent, stabilize and perhaps regress coronary heart diseases through their direct action on the vascular wall. This includes correction of endothelial dysfunction (vasoreactivity, monocyte adhesion, haemostasis), improvement of plaque stability (decrease in MMP expression), reduction of monocyte infiltration, decrease in oxidation level, decrease in cell proliferation, and repair of ischemic tissues through the mobilization of endothelial progenitors. These pleiotropic effects are explained by the fact that statins inhibit the intracellular production of metabolites located downstream of mevalonate in the cholesterol pathway, such as noids (farnesy pyrophosphate and geranylgeranylpyrophosphate). These hydrophobic metabolites allow the membrane anchorage of small G proteins (Ras and Rho) as well as the Gγ subunit of heterotrimeric G proteins, a post-translational step that is critical in the regulation of G protein signalling activity. Accordingly, the alteration of gene expression induced by statins can be reversed upon addition of these isoprenoids and not by cholesterol. The beneficial effects of statins are not restricted to the cardiovascular system; bone regeneration, cancer and HIV infection are also targeted. These drugs are therefore a promising tool, not only for the clinician but also for the biologist, allowing him/her to investigate the regulation of the gene expression that is controlled by the intracellular activity of membrane-anchored prenylated signalling proteins.
© 2002 médecine/sciences - Inserm / SRMS
L’athérosclérose est une maladie inflammatoire chronique, lentement évolutive, qui affecte la morphologie et la fonction des artères au niveau de zones bien précises, en particulier les bifurcations où l’écoulement sanguin est irrégulier. C’est une pathologie résultant d’étiologies multiples qui font intervenir des facteurs environnementaux, des anomalies plasmatiques et génétiques, ainsi que des dysfonctionnements cellulaires. Ainsi, une succession d’événements conduit à la formation du thrombus au niveau de la paroi, prélude à l’obstruction du vaisseau: infiltration puis oxydation des LDL (low density lipoproteins), pénétration des cellules immunitaires, production de médiateurs pathogènes (cytokines inflammatoires, radicaux libres oxygénés, chimiokines chimio-attractrices…), différenciation des monocytes en macrophages spumeux suivie de leur apoptose et donc de l’augmentation du cœur lipidique pariétal, dysfonctionnement endothélial, protéolyse péricellulaire favorisant la rupture de plaque… Ainsi, nombreuses sont les stratégies thérapeutiques qui, pour réduire l’incidence des pathologies isché-miques, ont pour objectif d’abaisser le taux de cholestérol associé aux LDL circulantes.
Dans un travail rassemblant les études d’intervention pharmacologique et nutritionnelle réalisées entre 1966 et 1996, Bucher et al. [1] concluent que les statines apportent une protection supérieure, en termes de décès d’origine cardiovasculaire ou autre, à celle des autres traitements hypolipémiants (fibrates, résines, acides gras n-3…). Les études cliniques de prévention secondaire (4S, CARE, LIPID) ou primaire (Woscops, AFCAPS/TexCAPS) des maladies cardiovasculaires rapportent des diminutions de la cholestérolémie totale de 25 % à 35 % après traitement par les statines, et l’on peut effectivement attendre d’un traitement hypolipémiant qu’il apporte à long terme un bénéfice lié à la diminution de l’infiltration pariétale de LDL potentiellement athérogènes. Cependant, des analyses par sousgroupes d’individus ont progressivement conduit cliniciens et biologistes à envisager que la baisse du cholestérol circulant observée lors d’un traitement par les statines n’était pas la seule responsable de la protection cardiovasculaire. D’ailleurs, le contexte inflammatoire dans lequel agissent les statines, la rapidité de leurs effets et leur mécanisme d’action intracellulaire confortent l’idée que ces molécules corrigent le dysfonctionnement vasculaire indépendamment de la baisse du cholestérol circulant.
Les effets secondaires indésirables des statines (douleurs musculaires, rhabdomyolyse) sont connus. En revanche, les décès associés à la prise de cérivastatine rapportés durant l’été 2001 semblent liés à des surdosages et à son association avec les fibrates. Toutefois, il n’est pas exclu que ces effets aient été aggravés par une toxicité propre à la molécule elle-même, ce qui a justifié son retrait du marché: en effet, de telles complications n’avaient encore jamais été décrites depuis que les statines sont prescrites. L’objectif ici n’est pas de débattre de ce sujet, car on ne dispose pas de tous les éléments d’appréciation nécessaires.
Statines : origine et mode d’action
Le premier des composés étudiés au début des années 1980 a été la mévastatine, isolée du champignon Penicillium citrinum. La simvastatine et la lovastatine sont également d’origine fongique (Aspergillus terreus), tandis que la pravastatine est obtenue après modification chimique de la bactérie Nocardia autotrophica. La fluvastatine et l’atorvastatine sont quant à elles synthétiques.
Les statines, du fait de leur analogie structurale avec l’HMG-CoA, inhibent de façon compétitive et réversible sa transformation en mévalonate par l’HMG-CoA réductase. Il s’ensuit une baisse de la concentration de tous les métabolites situés en aval du mévalonate, dont le cholestérol. Dans le foie, la diminution de la concentration intracellulaire en cholestérol entraîne une augmentation de l’expression des récepteurs hépatiques des LDL, ce qui conduit à un accroissement de l’épuration plasmatique des LDL circulantes. Ce phénomène, associé à une diminution de la production des VLDL (very low density lipoproteins) hépatiques et à une augmentation des HDL (high density lipoproteins) anti-athérogènes [2], améliore ainsi sensiblement le profil lipoprotéique. Les statines, en agissant très en amont dans la chaîne de synthèse du cholestérol, inhibent de ce fait la production de nombreux métabolites issus du mévalonate (Figure 1), et plus particulièrement des isoprénoïdes, farnésylpyrophosphate (FPP) et géranylgéranylpyrophosphate (GGPP). Ces composés interviennent dans la modification post-traductionnelle de différentes protéines de signalisation qui lient et hydrolysent le GTP. Ces protéines, qui ne possèdent pas de domaine transmembranaire, ont besoin d’acquérir un ancrage hydrophobe apporté par ces isoprénoïdes, qui se fixent de façon covalente à une cystéine située dans la région C-terminale appartenant au motif CAAX des petites protéines G de type Ras ou Rho/Rac. La formation de cette liaison est catalysée par des prényltransférases, spécifiques du FPP (farnésyl transférase) ou du GGPP (géranylgéranyl transférase type I). La spécificité est dictée par la nature de l’acide aminé X: si celui-ci est la méthionine, la sérine, l’alanine ou la glutamine, ce sera le FFP; si X est une leucine, ce sera le GGPP. Ainsi, les protéines Rho/Rac, qui contiennent le motif CAAX se terminant par X = leucine, seront prénylées par la géranylgéranyl transférase I.
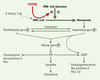 |
Figure 1. Action des statines sur la voie de synthèse du cholestérol et des isopré-noïdes. HMG-CoA: hydroxyméthylglutaryl-coenzyme A; FPP: farnésyl pyrophosphate; GGPP: géranylgéranyl pyrophosphate; PyroP: pyrophosphate. |
Si l’on prend l’exemple des protéines Ras, substrat des farnésyl transférases (Figure 2), la présence du résidu farnésyl accroît l’hydrophobicité de la protéine Ras et permet son ancrage à la membrane du réticulum endoplasmique. Une protéolyse spécifique destinée à ôter les acides aminés AAX ainsi qu’une étape de carboxyméthy-lation de la cystéine par une méthyl transférase spécifique des cystéines prénylées permettent de renforcer l’hydrophobicité de la protéine Ras prénylée et de stabiliser son accrochage à la membrane. Cette séquence de modifications post-traductionnelles est essentielle dans l’adressage des petites protéines G à la membrane plasmique et participe de façon décisive à la régulation de leur activité de signalisation. Ainsi, l’inhibition de la synthèse des isoprénoïdes par les statines diminue le niveau de prénylation des protéines G et, par voie de conséquence, la quantité de ces protéines ancrées à la membrane, et donc actives. Soulignons que la sous-unité ydes protéines G hétérotrimériques est également prénylée par le GGPP, ce qui affecte notamment l’activité des sous-unités effectrices αi et αs.
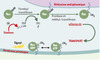 |
Figure 2. Modification post-traductionnelle des petites protéines G par les iso-prénoïdes: exemple de Ras. La farnésyl transférase (FT) catalyse le transfert du farnésyl pyrophosphate (FPP) sur la cystéine appartenant au motif CAAX. Cette liaison permet l’accrochage de Ras à la membrane du réticulum endoplasmique. Cet accrochage est renforcé par la coupure protéolytique (CAAX protéase) suivie d’une méthylation par une méthyl transférase dépendante de la cystéine prénylée. L’ensemble est transporté dans des vésicules golgiennes à la membrane plasmique où Ras pourra exercer, grâce à son activité d’échange et d’hydrolyse du GTP/GDP, sa fonction de signalisation intracellulaire. Les statines, en réduisant l’apport en FPP, perturbent de ce fait l’étape cruciale, initiale, d’accrochage de Ras. |
On imagine aisément que le spectre d’action des statines est large, puisque les protéines G, placées en amont des voies de signalisation, participent à la régu-ation de l’expression de nombreux gènes et fonctions cellulaires.
Statines et tonus vasculaire
Le dysfonctionnement endothélial est l’une des caractéristiques majeures de la pathologie athéroscléreuse. Elle se manifeste en particulier par une perte de la vasoreaxation, que l’on peut évaluer par la réponse à l’acétylcholine du débit sanguin au niveau de l’avant-bras (plé-thysmographie). Chez les patients présentant une hypercholestérolémie sévère, les statines améliorent les propriétés vasodilatatrices de l’endothélium [3]. Ces améliorations sont rapides, puisqu’elles sont observées après un mois seulement de prise du médicament, et sont dissociées de la baisse du taux de cholestérol circulant [4].
Pour tenter d’expliquer ces effets, les chercheurs se sont logiquement intéressés à la modulation de la production du monoxyde d’azote (NO), médiateur clé du tonus vasculaire. La production de NO est neutralisée par les LDL oxydées qui inhibent la transcription du gène de la NO synthase endothéliale. Dans les cellules endothéliales en culture, l’inhibition de la NO synthase par les LDL oxydées est levée par l’atorvastatine, qui restaure des niveaux normaux d’ARNm de la protéine en les stabilisant [5]. L’effet de la statine est bien lié à l’inhibition de l’HMG-CoA réductase, puisque l’ajout de mévalonate, en rétablissant la voie de synthèse des isoprénoïdes, annule cet effet. Ces résultats concordent avec ceux obtenus in vivo, où la biodisponibilité du NO, abaissée chez les sujets hypercholestérolémiques, est rétablie par un traitement à base de satines [3]. Un mécanisme complémentaire d’action des statines contribuerait à augmenter la biodisponibilité du NO, via une inhibition de la production de l’anion superoxyde produit par la NAD(P)H oxydase. Les statines, en empêchant l’induction de cette enzyme par les LDL oxydées, réduiraient la production d’anion superoxyde et de ce fait l’inactivation de NO [6].
Les statines diminuent la vasoconstriction en réduisant la production d’endothéline par l’intermédiaire d’une baisse significative des niveaux de l’ARNm du précurseur de l’endothéline (pré-proendothéline) [7]. Le mécanisme implique l’inhibition de l’activité de la protéine Rho par défaut de prénylation, puisque l’ajout de GGPP annule l’effet de la statine.
Statines et contexte inflammatoire
On sait que les médiateurs de l’inflammation jouent un rôle primordial dans le développement de l’athérosclérose [8]. La protéine C réactive (CRP) a longtemps été considérée comme un marqueur de l’état inflammatoire accompagnant l’athérosclérose. On semble lui accorder aujourd’hui un rôle plus direct dans cette pathologie, à travers ses effets proinflammatoires: stimulation de la synthèse du facteur tissulaire (FT) et du facteur chimio-attracteur des monocytes, MCP-1. Les travaux de Ridker et al. ont montré que le contexte inflammatoire que révèle la CRP, en particulier, serait une composante déterminante de l’efficacité des statines. Dans une étude princeps [9], ces auteurs ont en effet évalué l’impact d’un traitement par la pravastatine sur le risque de récidive d’infarctus chez des sujets préalablement classés en quintiles croissants de concentration sérique en CRP. Il a été constaté que la statine exerçait son effet protecteur le plus prononcé, en termes de récidive, dans le quintile où le taux de CRP était le plus élevé. Cette action semblait indépendante de la cholestérolémie, puisque celle-ci était, au début de l’étude, comparable chez les sujets présentant ou non des signes d’inflammation. De même, chez des sujets coronariens présentant des risques élevés d’infarctus du myocarde, le traitement par la statine s’est avéré plus protecteur, en termes de survie, chez les patients ayant le taux de CRP le plus élevé [10]. Cet effet protecteur est concordant avec le fait que les statines réduisent les niveaux de CRP circulante [11].
Statines et potentiel fibrinolytique endothélial
Les statines moduleraient également certaines propriétés hémostatiques de l’endothélium. La diminution de la thrombomoduline, marqueur de la souffrance endothéliale, observée chez les transplantés cardiaques traités par la fluvastatine en est ainsi le reflet [12]. Le PAI-1, inhibiteur des activateurs du plasminogène (t-PA et u-PA) de type I, ralentit la dégradation du caillot de fibrine par la plasmine. Il est à ce titre considéré comme potentiellement impliqué dans le risque thrombotique, des taux élevés de PAI-1 circulant constituant un facteur de risque d’infarctus du myocarde, en particulier chez les sujets obèses résistants à l’insuline [13]. Le PAI-1 interviendrait également dans le développement de lésions vasculaires, même si la nature de son rôle reste controversée. En effet, le PAI-1, en empêchant la formation de plasmine, a une action négative sur l’activation des métalloprotéases, et peut donc diminuer le risque de rupture de la plaque d’athérosclérose. En revanche, il peut également favoriser les dépôts de fibrine intrapariétaux et contribuer ainsi à maintenir un état inflammatoire, et donc la croissance de la plaque [14].
Les résultats des études cliniques réalisées jusqu’à présent ne permettent pas de conclure à un effet univoque des statines sur le potentiel fibrinolytique plasmatique. Cependant, la mesure du taux circulant du PAI-1 n’est pas le reflet fidèle de celui existant au niveau des zones lésées de la paroi, mais de l’ensemble des territoires dans lesquels il est produit (tissu adipeux, foie, arbre vasculaire). Différents résultats obtenus sur des cultures de cellules endothéliales humaines indiquent que les statines ont tendance à augmenter le potentiel fibri-nolytique de la cellule endothéliale en diminuant la production de PAI-1 et en augmentant celle du t-PA [15, 16]. L’effet inhibiteur sur la synthèse de PAI-1 est de type transcriptionnel et implique probablement la protéine Rho, puisque l’ajout du GGPP empêche cette inhibition. Il est donc possible que les statines concourent à imiter l’accumulation de fibrine au niveau de zones de la paroi (ulcération, apoptose) propices à l’activation de la coagulation.
Statines et potentiel d’adhérence endothélial
L’adhérence des monocytes à l’endothélium, processus de type inflammatoire, est l’une des étapes les plus précoces de l’athérosclérose. En effet, dès que les LDL ont infiltré l’intima et subi une oxydation même minime, elles induisent dans la cellule endothéliale l’expression de molécules d’adhérence (E-sélectine, P-sélectine, VCAM-1 - vascular cell adhésion molécule, ICAM-1 -intercellular adhésion molécule) qui reconnaissent leurs igands monocytaires, les intégrines. Les monocytes infiltrent ensuite la paroi vers les zones de l’intima où siège le foyer inflammatoire lésionnel. Si ce processus est salvateur au début, puisque chargé d’éliminer des composants athérogènes dans la paroi (LDL oxydées en particulier), il devient progressivement pathogène en raison d’un dérèglement du macrophage.
La technique de microscopie intravitale a permis de montrer que, chez le rat hypercholestérolémique, les statines entraînent une baisse de l’adhérence des monocytes à l’endothélium induite par des inhibiteurs de production de NO ou par le PAF (platelet activating factor) [17]. Ce phénomène s’explique bien au niveau du monocyte, puisque les statines y diminuent l’expression de surface des intégrines [18]. De plus, une autre propriété originale des statines, indépendante de la voie du mévalonate, est de reconnaître un domaine extracelulaire de l’intégrine LFA-1 (leukocyte function antigen-l) et d’empêcher ainsi l’adhérence cellulaire. Des modifications ciblées de la molécule de statine rendant saiaison à l’intégrine plus spécifique ont confirmé ses propriétés anti-inflammatoires chez la souris [19].
En revanche, l’effet des statines au niveau de la cellule endothéliale semble plus complexe. L’adhérence des monocytes à la cellule endothéliale stimulée par le TNFα (tumor necrosis factor α ), mesurée in vitro en conditions de flux, est significativement diminuée par l’ator-vastatine. De façon surprenante, on constate une augmentation de l’expression des molécules d’adhérence (ICAM-1, VCAM-1 et E-sélectine) à la surface de la cellule endothéliale[20] , au lieu de la diminution logiquement attendue, telle qu’elle est observée, par exemple, dans le monocyte.
Comment expliquer ce paradoxe? Le regroupement (clustering) des molécules d’adhérence est indispensable à leur fonction d’adhérence, et le rôle clé de Rho dans ce processus a été proposé [21]. Or, l’ajout de GGPP rétablit le potentiel d’adhérence endothélial à un niveau proche de celui induit par le TNFα seul, et la distribution des molécules d’adhérence en surface, observée en microscopie confocale, est modifiée par la statine [20]. Ainsi, l’hypothèse formulée est que la désorganisation des molécules d’adhérence liée à l’inactivation de Rho par la statine entraînerait une moins bonne reconnaissance du monocyte par la cellule endothéliale, et ce indépendamment du niveau d’expression des molécules d’adhérence à la surface cellulaire (Figure 3). La potentialisation de l’expression de surface des molécules d’adhérence par la statine est le résultat d’une augmentation de leur synthèse (ARNm et protéine), dont les mécanismes sont actuellement à l’étude. Ces résultats soulignent la grande complexité d’action des statines puisqu’elles peuvent moduler à la fois l’expression et la fonction de certaines protéines, ici les molécules d’adhérence.
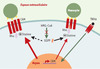 |
Figure 3. Mécanismes hypothétiques de l’effet des statines dans la cellule endothé-liale et de leurs conséquences sur l’adhérence des monocytes. Les molécules d’adhérence (CAM) (en rouge) sont exprimées à la surface de la cellule endothéliale en réponse au TNFα . La protéine Rho est correctement ancrée à la membrane grâce au GGPP et, en interaction avec le cytosquelette, assure le regroupement fonctionnel des molécules d’adhérence, ce qui permet l’adhérence optimale des monocytes (à droite sur la figure, voir [21]). En empêchant la prénylation de Rho par le GGPP, les statines (à gauche) perturbent de ce fait le regroupement des CAM endothéliales, donc l’adhérence. L’effet activateur des statines sur le niveau d’expression des CAM induit par le TNFα est représenté par un nombre plus élevé de barres. |
Statines et stabilité de la plaque d’athérosclérose
La stabilité de la plaque dépend plus de sa composition que de sa taille. Une plaque riche en lipides et pauvre en collagène, donc peu fibreuse, présente des risques importants de fissuration, sans pour autant que l’imagerie ait pu détecter un rétrécissement significatif de la lumière du vaisseau. Parmi les facteurs d’instabilité, l’activité des métalloprotéases matricielles (MMP) joue un rôle certain. Ces enzymes sont libérées par les macrophages et participent au remodelage tissulaire en dégradant la matrice extracellulaire. Produites et activées en excès, elles contribuent à fragiliser la paroi, et c’est effectivement dans les zones de rupture potentielle riches en macrophages que cette activité est la plus importante. Les interventions médicamenteuses visant à stabiliser la plaque sont donc d’un intérêt thérapeutique majeur, et il semble que l’on puisse attribuer aux statines de telles propriétés.
Un traitement de 3 mois par la pravastatine entraîne une baisse du contenu en lipides (et de leur oxydation) des plaques carotidiennes humaines, analysées après endartériectomie, une diminution de l’apoptose et de l’activité MMP-2, ainsi qu’une augmentation du contenu en collagène [22]. Grâce aux progrès importants réalisés dans le domaine de l’imagerie (IRM en particulier), des données concernant la structure de la plaque sont désormais accessibles de façon non invasive, et vont certainement permettre dans les prochaines années une meilleure évaluation des risques. Ainsi, l’IRM a permis de montrer que les statines, seules [23] ou en association avec d’autres hypolipémiants [24], font à long terme régresser la plaque et changent sa composition en faveur d’un meilleur pronostic de stabilité. Ces résultats vont dans le sens de ceux obtenus chez le singe [25] et le lapin [26], chez qui les statines ralentissent sensiblement la progression de la plaque, évaluée par la mesure de différents paramètres tels que l’épaisseur intima-média (EIM), le dépôt de collagène ou la quantité de lipides d’origine macrophagique. Dans l’étude chez le singe, l’indépendance de ce phénomène vis-à-vis du taux de cholestérol circulant est soulignée grâce à l’ajustement de protocoles nutritionnels permettant de normaliser les taux de cholestérol entre les groupes traités ou non par la statine. Les auteurs ont constaté que l’EIM, la calcification de la plaque et l’infiltration macrophagique étaient plus faibles dans les groupes traités par la statine, et s’accompagnaient d’une amélioration de la réactivité vasculaire à l’acétylcholine. De même, l’étude CAIUS (carotid atherosclerosis Italian ultrasound study) [27] montre une stabilisation de l’EIM quelle que soit la baisse du cholestérol en réponse à la pravastatine, suggérant une dissociation entre la baisse du cholestérol et l’amélioration des paramètres de stabilité de la plaque.
Deux domaines au moins peuvent être repérés, dans lesquels des modifications cellulaires et fonctionnelles pourraient rendre compte des effets bénéfiques des statines sur la paroi.
Au niveau des monocytes/macrophages, outre le mécanisme d’inhibition de l’adhérence monocytaire à l’endothélium décrit plus haut, s’ajoute le fait que les statines inhibent la synthèse de la MCP-1. Protéine de l’inflammation, la MCP-1 favorise la migration intimale des monocytes. Chez la souris, l’administration de lovastatine ou de simvastatine diminue considérablement sa production [28]. Il s’agit d’une inhibition de type trans-criptionnel, qui est levée par l’ajout de dérivés du mévalonate. Une action inhibitrice des statines sur la migration intimale des monocytes est donc possible.
Dans les macrophages isolés en culture, la synthèse et l’activité de la MMP-9 sont diminuées par la fluvastatine [29]. Il en est de même in vivo chez le lapin athéroscléreux traité par la cérivastatine [30]. Ce phénomène pourrait contribuer à diminuer l’activité protéolytique, d’autant que la production de PAI-1 par le macrophage n’est pas diminuée par la statine [31]. Une action de stabilisation de la plaque par ce mécanisme est donc envisageable.
Chez l’homme, l’association mise en évidence entre la charge infectieuse et le risque cardiovasculaire suggère un rôle de l’infection dans le développement de l’athérosclérose. Les études en cours nous diront si effectivement la charge infectieuse est ou non un facteur prédisposant ou aggravant l’athérosclérose. Dans ce contexte, Kothe et al. montrent que les statines inhibent la réactivité des macrophages humains à l’infection par Chlamydia pneumoniae, ce qui se traduit par une diminution de la production de MCP-1 et d’IL-8 (interleukine-8), tandis que celle du TNFα reste inchangée [32].
Une autre composante du remodelage vasculaire sur laquelle les statines pourraient avoir un effet positif est la diminution des dépôts extravasculaires de fibrine. Dans un modèle d’athérosclérose induite chez le lapin, la cérivastatine réduit l’expression pariétale du facteur tissulaire [30]. Le facteur sur lequel agit la statine est d’origine macrophagique, ce qui est concordant avec les données obtenues dans des macrophages humains isolés, cellules dans lesquelles l’effet inhibiteur de la statine sur le FT serait lié à une diminution de l’activité du facteur de transcription NF-κB [33].
La cérivastatine diminue quant à elle l’expression, induite par les LDL oxydées, de l’u-PA et de son récepteur, ce qui pourrait moduler les propriétés d’adhérence des monocytes à la matrice protéique [34]. L’inhibition de la farnésylation de Ras et de la translocation de NF-κB rendrait compte de ces effets.
L’influence bénéfique des cellules musculaires lisses (CML) sur la stabilité de la plaque est maintenant bien admise, puisqu’en colonisant la chape fibreuse recouvrant la plaque, elles contribuent à la renforcer grâce en particulier au collagène qu’elles sécrètent. L’action des statines sur les CML est relativement complexe, car elle semble dépendre des propriétés lipophiles ou hydrophiles de la molécule. Cet aspect est illustré dans une étude chez le lapin qui montre que la pravastatine (hydrophile) accroît, dans les zones athéromateuses, la quantité de collagène (ARNm et protéine) par rapport aux animaux non traités [35], inhibant l’apoptose des CML, ce qui assure le maintien de cette population. En revanche, la fluvastatine (lipophile) ne modifie pas le contenu en collagène des plaques chez les animaux traités, et ne semble pas non plus empêcher l’apoptose des CML. Cela est confirmé in vitro dans des cultures de CML de rat. Les sta-tines lipophiles (lovastatine, fluvastatine et atorvasta-tine) provoquent l’apoptose, au contraire de la pravasta-tine [36]. Cette action pro-apoptotique implique la géra-nylgéranylation de la petite protéine G RhoB. L’effet anti-prolifératif des statines lipophiles a été décrit in vitro sur des CML stimulées par le PDGF (platelet derived growth factor) [37]. Or, l’action proliférative du PDGF est due à l’inhibition qu’il exerce sur l’activité de l’inhibiteur des Cdk (cyclin dependent kinase) p27kip1, par un mécanisme impliquant l’activité de Rho. En inhibant la prénylation de Rho, et donc sa translocation membranaire, la statine entraînerait sa perte d’activité et lèverait ainsi son pouvoir inhibiteur sur la p27kip1, ce qui conduirait à l’arrêt du cycle. En effet, le GGPP, mais pas le FPP, est capable d’empêcher l’arrêt de la prolifération des CML induit par la sta-tine. Les mécanismes responsables des différences d’effet entre les statines lipophiles et hydrophiles sur la prolifération des CML ne sont pas encore élucidés, ce qui souligne à nouveau la grande complexité d’action des sta-tines au niveau intracellulaire.
Soulignons également les résultats récents montrant que la synthèse de COX-2 et la production de prostacycline sont augmentées dans les CML aortiques humaines en culture sous l’action de la lovastatine [38]. Cette action contribuerait à l’amélioration de la vasoréactivité observée chez les sujets coronariens traités par les statines.
Statines et néo-vascularisation
Le rôle des progéniteurs de cellules endothéliales CD34+ issus de la moelle osseuse semble majeur dans la survie des zones ischémiées du myocarde. En colonisant les zones ischémiées, ils participent à la formation de néovaisseaux, ce qui rendrait compte de leurs effets positifs sur la fonction contractile et l’inhibition de l’apoptose [39]. Or, des travaux récents montrent que, chez des patients coronariens, l’atorvastatine augmente la quantité circulante de progéniteurs CD34+ acquérant une fonctionnalité migratoire [40]. Le processus facilitant la néo-vascularisation a également été décrit chez la souris avec la simvastatine; il implique la modulation de la voie phosphatidylinositol-3 kinase (PI3K)/Akt [41]. Cette voie, d’une part, module l’angiogenèse, via la production de NO par la NO synthase et, d’autre part, inhibe l’apoptose. La PI3K, par le PI3 qu’elle engendre, permet à la kinase dépendante des phospho-inositides (PDK-1) d’activer la protéine Akt. Or, l’activité de la PI3K est contrôlée négativement par un des métabolites en aval du mévalonate. Ainsi, en inhibant la synthèse de ces métabolites, la statine libère l’activité de la PI3K et permet l’activation d’Akt. Cependant, le mécanisme exact par lequel un des métabolites de la voie de synthèse du cholestérol inhibe la PI3K n’est pas encore élucidé. Il est néanmoins tentant de rapprocher ces données de celles obtenues chez la souris, où le prétraitement par la simvastatine avant induction de l’ischémie myocardique améliore sensiblement les paramètres de la fonction contractile du cœur, en comparaison avec les animaux non traités [42].
Conclusions
Ces quelques exemples montrent que le potentiel thérapeutique des statines s’exerce bien au-delà de la simple diminution du cholestérol LDL circulant, puisqu’elles corrigent le dysfonctionnement de la paroi vasculaire athéroscléreuse en agissant sur les régulations fonctionnelles et sécrétoires de ses principaux constituants cellulaires: cellule endothéliale, cellule musculaire lisse et macrophage (Figure 4).
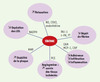 |
Figure 4. Effets pléiotropes vasculaires des statines. PAI-1: inhibiteur des acti-vateurs du plasminogène de type I; t-PA: activateur du plasminogène tissu-laire; FT: facteur tissulaire ; CAM : cellular adhesion molecules ; MCP-1: facteur chimio-attracteur des monocytes; CRP: protéine C réactive; PCE: progéniteurs de cellules endothéliales ; MMP: métalloprotéases matricielles. |
Les statines sont à l’étude dans d’autres maladies comme l’ostéoporose, certains cancers et aussi l’infection par le VIH. Dans ce dernier cas, certaines régions spécifiques de la membrane, riches en cholestérol, les rafts, seraient des zones de reconnaissance et de pénétration privilégiée du VIH. Les statines, en diminuant les teneurs en cholestérol dans ces régions, pourraient de cette manière moduler négativement la fixation du virus et donc l’infection [43].
Alors, sommes nous en présence d’une classe surprenante de molécules, un peu à la manière de l’aspirine du siècle dernier? L’avenir nous le dira.
Références
- Bucher HC, Griffith LE, Guyatt GH. Systematic review on the risk and benefit of different cholesterol lowering interventions. Arterioscler Thromb Vasc Biol 1999; 19: 187–95. [Google Scholar]
- Martin G, Duez H, Blanquart C, et al. Statin-induced inhibition of the Rho-signaling pathway activates PPARalpha and induces HDL apoA-I. J Clin Invest 2001; 107: 1423–32. [Google Scholar]
- John S, Schlaich M, Langenfeld M, et al. Increased bioavailability of nitric oxide after lipid-lowering therapy in hypercholesterolemic patients: a randomized, placebo-controlled, double-blind study. Circulation 1998; 98: 211–6. [Google Scholar]
- O’Driscoll G, Green D, Taylor RR. Simvastatin, an HMG-coenzyme A reductase inhibitor, improves endothelial function within 1 month. Circulation 1997; 95: 1126–31. [Google Scholar]
- Hernandez-Perera O, Perez-Sala D, Navarro-Antolin J, et al. Effects of the 3-hydroxy-3-methylglutaryl-CoA reductase inhibitors, atorvastatin and simvastatin, on the expression of endothelin-1 and endothelial nitric oxide synthase in vascular endothelial cells. J Clin Invest 1998; 101: 2711–9. [Google Scholar]
- Rueckschloss U, Galle J, Holtz J, et al. Induction of NAD (P) H oxidase by oxidized low-density lipoprotein in human endothelial cells: antioxidative potential of hydroxymethylglutaryl coenzyme A reductase inhibitor therapy. Circulation 2001; 104: 1767–72. [Google Scholar]
- Hernandez-Perera O, Perez-Sala D, Soria E, Lamas S. Involvement of rho GTPases in the transcriptional inhibition of preproendothelin-1 gene expression by simvastatin in vascular endothelial cells. Circ Res 2000; 87: 616–22. [Google Scholar]
- Tedgui A, Mallat Z. Athérosclérose et inflammation. Med Sci 2001; 17: 162–9. [Google Scholar]
- Ridker PM, Rifai N, Pfeffer MA, et al. Inflammation, pravastatin, and the risk of coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and recurrent events (CARE) investigators. Circulation 1998; 98: 839–44. [Google Scholar]
- Horne BD, Muhlestein JB, Carlquist JF, et al. Statin therapy, lipid levels, C-reactive protein and the survival of patients with angiographically severe coronary artery disease. J Am Coll Cardiol 2000; 36: 1774–80. [Google Scholar]
- Ridker PM, Rifai N, Pfeffer MA, Sacks F, Braunwald E. Long-term effects of pravastatin on plasma concentration of C-reactive protein. Circulation 1999; 100: 230–5. [Google Scholar]
- Ambrosi P, Aillaud MF, Habib G, et al. Fluvastatin decreases soluble thrombomodulin in cardiac transplant recipients. Thromb Haemost 2000; 83: 46–8. [Google Scholar]
- Juhan-Vague I, Alessi MC. Regulation of fibrinolysis in the development of atherothrombosis: role of adipose tissue. Thromb Haemost 1999; 82: 832–6. [Google Scholar]
- Nalbone G, Alessi MC, Juhan-Vague I. Système fibrinolytique, métalloprotéases et pathologie vasculaire. Med Sci 2001; 17: 170–6. [Google Scholar]
- Essig M, Nguyen G, Prie D, Escoubet B, Sraer JD, Friedlander G. 3-Hydroxy-3-methylglutaryl coenzyme A reductase inhibitors increase fibrinolytic activity in rat aortic endothelial cells. Role of geranylgeranylation and Rho proteins. Circ Res 1998; 83: 683–90. [Google Scholar]
- Lopez S, Peiretti F, Bonardo B, Juhan-Vague I, Nalbone G. Effect of atorvastatin and fluvastatin on the expression of plasminogen activator inhibitor type-1 in cultured human endothelial cells. Atherosclerosis 2000; 152: 359–66. [Google Scholar]
- Pruefer D, Scalia R, Lefer AM. Simvastatin inhibits leukocyte-endothelial cell interactions and protects against inflammatory processes in normocholesterolemic rats. Arterioscler Thromb Vasc Biol 1999; 19: 2894–900. [Google Scholar]
- Yoshida M, Sawada T, Ishii H, et al. HMG-CoA reductase inhibitor modulates monocyte-endothelial cell interaction under physiological flow conditions in vitro: involvement of Rho GTPase-dependent mechanism. Arterioscler Thromb Vasc Biol 2001; 21: 1165–71. [Google Scholar]
- Weitz-Schmidt G, Welzenbach K, Brinkmann V, et al. Statins selectively inhibit leucocyte function antigen-1 by binding to a novel regulatory integrin site. Nat Med 2001; 7 : 687–92. [Google Scholar]
- Bernot D, Benoliel AM, Peiretti F, et al. Atorvastatin inhibits monocyte adhesion to TNFα-activated human endothelial cells (HUVEC). J Cardiovasc Pharmacol 2003 (sous presse). [Google Scholar]
- Wojciak-Stothard B, Williams L, Ridley AJ. Monocyte adhesion and spreading on human endothelial cells is dependent on Rho-regulated receptor clustering. J Cell Biol 1999; 145: 1293–307. [Google Scholar]
- Crisby M, Nordin-Fredriksson G, Shah PK, et al. Pravastatin treatment increases collagen content and decreases lipid content, inflammation, metalloproteinases, and cell death in human carotid plaques: implications for plaque stabilization. Circulation 2001; 103: 926–33. [Google Scholar]
- Corti R, Fayad ZA, Fuster V, et al. Effects of lipid-lowering by simvastatin on human atherosclerotic lesions: a longitudinal study by high-resolution, noninvasive magnetic resonance imaging. Circulation 2001; 104: 249–52. [Google Scholar]
- Zhao XQ, Yuan C, Hatsukami TS, et al. Effects of prolonged intensive lipid-lowering therapy on the characteristics of carotid atherosclerotic plaques in vivo by MRI: a case-control study. Arterioscler Thromb Vasc Biol 2001; 21: 1623–9. [Google Scholar]
- Williams JK, Sukhova GK, Herrington DM, Libby P. Pravastatin has cholesterol-lowering independent effects on the artery wall of atherosclerotic monkeys. J Am Coll Cardiol 1998; 31: 684–91. [Google Scholar]
- Bustos C, Hernandez-Presa MA, Ortego M, et al. HMG-CoA reductase inhibition by atorvastatin reduces neointimal inflammation in a rabbit model of atherosclerosis. J Am Coll Cardiol 1998; 32: 2057–64. [Google Scholar]
- Baldassarre D, Veglia F, Gobbi C, et al. Intima-media thickness after pravastatin stabilizes also in patients with moderate to no reduction in LDL-cholesterol levels: the carotid atherosclerosis italian ultrasound study. Atherosclerosis 2000; 151: 575–83. [Google Scholar]
- Romano M, Diomede L, Sironi M, et al. Inhibition of monocyte chemotactic protein-1 synthesis by statins. Lab Invest 2000; 80: 1095–100. [Google Scholar]
- Bellosta S, Via D, Canavesi M, et al. HMG-CoA reductase inhibitors reduce MMP-9 secretion by macrophages. Arterioscler Thromb Vasc Biol 1998; 18: 1671–8. [Google Scholar]
- Aikawa M, Rabkin E, Sugiyama S, et al. An HMG-CoA reductase inhibitor, cerivastatin, suppresses growth of macrophages expressing matrix metalloproteinases and tissue factor in vivo and in vitro. Circulation 2001; 103: 276–83. [Google Scholar]
- Lopez S, Peiretti F, Bonardo B, et al. Effect of atorvastatin on plasminogen activator inhibitor type-1 synthesis in human monocytes/macrophages. J Cardiovasc Pharmacol 2001; 37: 762–8. [Google Scholar]
- Kothe H, Dalhoff K, Rupp J, et al. Hydroxymethylglutaryl coenzyme a reductase inhibitors modify the inflammatory response of human macrophages and endothelial cells infected with Chlamydia pneumoniae. Circulation 2000; 101: 1760–3. [Google Scholar]
- Colli S, Eligini S, Lalli M, et al. Vastatins inhibit tissue factor in cultured human macrophages. A novel mechanism of protection against atherothrombosis. Arterioscler Thromb Vasc Biol 1997; 17: 265–72. [Google Scholar]
- Ganne F, Vasse M, Beaudeux JL, et al. Cerivastatin, an inhibitor of HMG-CoA reductase, inhibits urokinase/urokinase-receptor expression and MMP-9 secretion by peripheral blood monocytes : a possible protective mechanism against atherothrombosis. Thromb Haemost 2000; 84: 680–8. [Google Scholar]
- Fukumoto Y, Libby P, Rabkin E, et al. Statins alter smooth muscle cell accumulation and collagen content in established atheroma of watanabe heritable hyperlipidemic rabbits. Circulation 2001; 103: 993–9. [Google Scholar]
- Guijarro C, Blanco-Colio LM, Massy ZA, et al. Lipophilic statins induce apoptosis of human vascular smooth muscle cells. Kidney Int 1999; 71 (suppl): S88–91. [Google Scholar]
- Laufs U, Marra D, Node K, Liao JK. 3-hydroxy-3-methylglutaryl-CoA reductase inhibitors attenuate vascular smooth muscle proliferation by preventing rho GTPase-induced down-regulation of p27 (Kip1). J Biol Chem 1999; 274: 21926–31. [Google Scholar]
- Degraeve F, Bolla M, Blaie S, et al. Modulation of COX-2 expression by statins in human aortic smooth muscle cells: involvement of geranylgeranylated proteins. J Biol Chem 2001; 276: 46849–55. [Google Scholar]
- Orlic D, Kajstura J, Chimenti S, et al. Mobilized bone marrow cells repair the infarcted heart, improving function and survival. Proc Natl Acad Sci USA 2001; 98: 10344–9. [Google Scholar]
- Vasa M, Fichtlscherer S, Adler K, et al. Increase in circulating endothelial progenitor cells by statin therapy in patients with stable coronary artery disease. Circulation 2001; 103: 2885–90. [Google Scholar]
- Llevadot J, Murasawa S, Kureishi Y, et al. HMG-CoA reductase inhibitor mobilizes bone marrow-derived endothelial progenitor cells. J Clin Invest 2001; 108: 399–405. [Google Scholar]
- Jones S P, Trocha SD, Lefer DJ. Pretreatment with simvastatin attenuates myocardial dysfunction after ischemia and chronic reperfusion. Arterioscler Thromb Vasc Biol 2001; 21: 2059–64. [Google Scholar]
- Ono A, Freed EO. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc Natl Acad Sci USA 2001; 98: 13925–30. [Google Scholar]
Liste des figures
|
Figure 1. Action des statines sur la voie de synthèse du cholestérol et des isopré-noïdes. HMG-CoA: hydroxyméthylglutaryl-coenzyme A; FPP: farnésyl pyrophosphate; GGPP: géranylgéranyl pyrophosphate; PyroP: pyrophosphate. |
| Dans le texte | |
|
Figure 2. Modification post-traductionnelle des petites protéines G par les iso-prénoïdes: exemple de Ras. La farnésyl transférase (FT) catalyse le transfert du farnésyl pyrophosphate (FPP) sur la cystéine appartenant au motif CAAX. Cette liaison permet l’accrochage de Ras à la membrane du réticulum endoplasmique. Cet accrochage est renforcé par la coupure protéolytique (CAAX protéase) suivie d’une méthylation par une méthyl transférase dépendante de la cystéine prénylée. L’ensemble est transporté dans des vésicules golgiennes à la membrane plasmique où Ras pourra exercer, grâce à son activité d’échange et d’hydrolyse du GTP/GDP, sa fonction de signalisation intracellulaire. Les statines, en réduisant l’apport en FPP, perturbent de ce fait l’étape cruciale, initiale, d’accrochage de Ras. |
| Dans le texte | |
|
Figure 3. Mécanismes hypothétiques de l’effet des statines dans la cellule endothé-liale et de leurs conséquences sur l’adhérence des monocytes. Les molécules d’adhérence (CAM) (en rouge) sont exprimées à la surface de la cellule endothéliale en réponse au TNFα . La protéine Rho est correctement ancrée à la membrane grâce au GGPP et, en interaction avec le cytosquelette, assure le regroupement fonctionnel des molécules d’adhérence, ce qui permet l’adhérence optimale des monocytes (à droite sur la figure, voir [21]). En empêchant la prénylation de Rho par le GGPP, les statines (à gauche) perturbent de ce fait le regroupement des CAM endothéliales, donc l’adhérence. L’effet activateur des statines sur le niveau d’expression des CAM induit par le TNFα est représenté par un nombre plus élevé de barres. |
| Dans le texte | |
|
Figure 4. Effets pléiotropes vasculaires des statines. PAI-1: inhibiteur des acti-vateurs du plasminogène de type I; t-PA: activateur du plasminogène tissu-laire; FT: facteur tissulaire ; CAM : cellular adhesion molecules ; MCP-1: facteur chimio-attracteur des monocytes; CRP: protéine C réactive; PCE: progéniteurs de cellules endothéliales ; MMP: métalloprotéases matricielles. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.