")
")
| Issue |
Med Sci (Paris)
Volume 18, Number 6-7, Juin–Juillet 2002
|
|
|---|---|---|
| Page(s) | 659 - 661 | |
| Section | Le Magazine : Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/20021867659 | |
| Published online | 15 juin 2002 | |
Une nouvelle classe de molécules hypolipémiantes : les ligands de SCAP
A new family of drugs lowering LDL
Laboratoire GlaxoSmithKline, 91951 Les Ulis Cedex, France
Une élévation du taux de LDL (low density lipoproteins)-cholestérol est un facteur de risque bien établi de maladies car-diovasculaires comme l’infarctus du myocarde. L’introduction des statines, une classe de médicaments qui inhibent la synthèse du cholestérol, dans le traitement des dyslipidémies a clairement démontré qu’une diminution du LDL-cholestérol se traduisait par une diminution de l’incidence des accidents cardiovasculaires et de la mortalité qu’ils entraînent [1]. Une relation mathématique a même été observée : les pourcentages de diminution du LDL-cholestérol, de la mortalité et de la morbidité secondaires à des accidents cardiovasculaires évoluent parallèlement. Les nouvelles générations de statines, de plus en plus puissantes, sont aussi de plus en plus efficaces, mais il semble qu’on atteigne une certaine limite au-delà de laquelle la toxicité apparaît. Cette toxicité s’oppose à l’escalade des doses de statines lorsque la réduction du LDL-cholestérol n’est pas suffisante. Les statines inhibent l’hydroxymétylglutaryl-Co-enzyme A réductase, une enzyme-clé de la synthèse du cholestérol. Si le cholestérol dans le plasma est délétère puisqu’il accélère le processus athérogène, il n’en demeure pas moins un constituant vital des cellules, impliqué dans de nombreux processus membranaires. Le récent retrait du marché de la cérivastatine souligne tout l’intérêt de la recherche de molécules capables de diminuer le LDL-cholestérol tout en épargnant le cholestérol cellulaire. Le foie, qui synthétise la grande majorité du cholestérol de l’organisme cellulaire et qui est le seul organe à produire des LDL, est essentiel à l’homéostasie du cholestérol. Qui plus est, il capte à nouveau la majorité des LDL circulantes via un récepteur spécifique, le LDL-récepteur [2], et produit les sels biliaires qui sont la principale voie d’élimination du cholestérol. Tous ces processus sont très précisément contrôlés, et les deux prix Nobel (→), Michael Brown et Joseph Goldstein en ont disséqué les bases moléculaires, notamment en identifiant une protéine baptisée SCAP (SREBP cleavage activating protein) et présentée comme le senseur du cholestérol [3–5] (→→). Lorsque les cellules sont appauvries en cholestérol, la protéine SCAP active la maturation de deux facteurs de transcription, SREBP-1α (sterol regulatory element-binding protein) et SREBP-2, qui activent à leur tour la transcription des gènes capables de restaurer le contenu en cholestérol des cellules (Figure 1A). Il s’agit principalement du LDL-récepteur et de l’hydroxymétylglutary-Co-enzyme A réductase, ce qui permet ainsi d’augmenter simultanément la synthèse de cholestérol et le captage des LDL-cholestérol à partir de la circulation. En établissant un test de criblage à haut débit utilisant une lignée hépatique humaine transfectée par une construction comprenant le promoteur du gène codant pour le récepteur des LDL couplé à un gène rapporteur, notre équipe de recherche a pu identifier une nouvelle classe de molécules capables d’activer la transcription du LDL-récepteur. Le mode d’action de ces molécules a été décortiqué et la cible moléculaire identifiée par photo-marquage d’affinité en utilisant un système d’expression dans le baculovirus et un analogue radiomarqué photo-activable. Ces molécules se fixent à un domaine de SCAP qui contient une séquence qui semble cruciale pour répondre à la concentration de cholestérol intracellulaire. Contrairement au cholestérol, qui bloque la maturation des SREBP, les ligands de SCAP que nous avons découverts stimulent la maturation des SREBP et activent ainsi la transcription du gène codant pour le LDL-récepteur. Nous avons localisé l’élément de réponse présent dans le promoteur de ce gène et qui est impliqué dans l’effet de nos molécules. Il correspond au sterol responsive element précédemment décrit par Brown et Goldstein [6]. D’une certaine manière les ligands de SCAP agissent comme des antagonistes de l’effet du cholestérol en mimant une déplétion en cholestérol. Bien que la liaison du cholestérol à SCAP n’ait jamais été démontrée, deux de nos molécules se fixent sur SCAP, et entrent en compétition pour ce site de fixation. Le schéma de la Figure 1B résume le mode d’action proposé des ligands de SCAP (noté GW).
(→) m/s 2000, n°4, p. 559
(→→) m/s 1985, n°7, p. 388
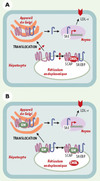 |
Figure 1. Régulation de l’expression du récepteur des LDL et action de l’inhibiteur (GW) de la protéine SCAP. A. Les protéines SCAP (violet) et SREBP (bleu) interagissent au niveau du réticulum endoplasmique pour former un complexe qui migre vers l’appareil de Golgi où SREBP subit un clivage protéolytique. La forme mature de SREBP migre vers le noyau pour interagir avec le sterol responsive element (SRE) présent sur le promoteur du récepteur des LDL, favorisant ainsi son expression. En présence d’un excès de cholestérol intracellulaire (chol), la translocation du complexe SCAP-SREBP est bloquée au niveau du réticulum endoplasmique (flèches rouges en A), inhibant ainsi l’expression du récepteur des LDL. B. Le composé GW se lie à SCAP, favorisant ainsi la translocation du complexe SCAP/SREBP même en présence d’un excès de cholestérol. |
Des hépatocytes primaires humains ou de primates traités par des ligands de SCAP sont capables d’internaliser 2 à 3 fois plus de LDL que des hépatocytes témoins. Cela confirme que l’augmentation de l’expression du LDL récepteur se traduit bien fonctionnellement par une plus grande capacité de capter les LDL. Dans un modèle animal, le hamster, les ligands de SCAP se sont montrés de puissants hypolipémiants. Des hamsters traités 3 jours par 5 mg/kg d’un ligand de SCAP réduisent leur concentration plasmatique de LDL-cholestérol de 64 %, alors que ni celle de HDL-cholestérol, ni celles des amino-transférases hépatiques ne sont modifiés, confirmant l’activité du ligand de SCAP, et son absence de toxicité. Les transcrits codant pour le LDL-récepteur sont augmentés d’environ trois fois dans les foies des hamsters traités. De manière intéressante, l’amplitude de l’effet hypocholestérolémiant est corrélée à la dose de ligand de SCAP administrée : cette amplitude varie de - 20 % pour une dose de 0,2 mg/kg pour atteindre - 80 % pour une dose de 20 mg/kg. Si les hamsters reçoivent une nourriture riche en lipides, une réduction des triglycérides d’au moins 60 % est observée dès la dose de 1 mg/kg [7]. L’amplitude de ces effets laisse penser que les ligands de SCAP pourraient représenter une nouvelle classe pharma-cologique d’hypolipémiants particulièrement prometteuse. Ces molécules ont démontré leur activité in vivo dans un modèle animal et in vitro sur des hépatocytes humains. Il sera donc nécessaire de confirmer leur activité chez l’homme avant d’envisager cette nouvelle classe pharmacologique comme une alternative thérapeutique.
Références
- Ansell BJ, Watson KE, Fogelman AM. An evidence-based assessment of the NCEP adult treatment panel II guidelines. National cholesterol education program. JAMA 1999; 282 : 2051–7. [Google Scholar]
- Kovanen PT, Schneider WJ. Regulation of the low density lipoprotein (B/E) receptor. Adv Vasc Biol 1999; 5 : 165–85. [Google Scholar]
- Hua X, Nohturfft A, Goldstein JL, Brown MS. Protein sterol resistance in CHO cells traced to point mutation in SREBP cleavage-activating protein. Cell 1996; 87 : 415–22. [Google Scholar]
- Matsuda M, Korn BS, Hammer RE, et al. SREBP cleavage-activating protein (SCAP) is required for increased lipid synthesis in liver induced by cholesterol deprivation and insulin elevation. Genes Dev 2001; 15 : 1206–16. [Google Scholar]
- Sakai J, Nohturfft A, Goldstein JL, Brown MS. Cleavage of sterol regulatory element-binding proteins (SREBPs) at site-1 requires interaction with SREBP cleavage-activating protein. Evidence from in vivo competition studies. J Biol Chem 1998; 273 : 5785–93. [Google Scholar]
- Briggs MR, Yokoyama C, Wang X, Brown MS, Goldstein JL. Nuclear protein that binds sterol regulatory element of low density lipoprotein receptor promoter. J Biol Chem 1993; 268 : 14490–6. [Google Scholar]
- Grand-Perret T, Bouillot A, Perrot A, Commans S, Walker M, Issandou M. SCAP ligands are potent new lipid-lowering drugs. Nat Med 2001; 7 :1332–8. [Google Scholar]
© 2002 médecine/sciences - Inserm / SRMS
Liste des figures
|
Figure 1. Régulation de l’expression du récepteur des LDL et action de l’inhibiteur (GW) de la protéine SCAP. A. Les protéines SCAP (violet) et SREBP (bleu) interagissent au niveau du réticulum endoplasmique pour former un complexe qui migre vers l’appareil de Golgi où SREBP subit un clivage protéolytique. La forme mature de SREBP migre vers le noyau pour interagir avec le sterol responsive element (SRE) présent sur le promoteur du récepteur des LDL, favorisant ainsi son expression. En présence d’un excès de cholestérol intracellulaire (chol), la translocation du complexe SCAP-SREBP est bloquée au niveau du réticulum endoplasmique (flèches rouges en A), inhibant ainsi l’expression du récepteur des LDL. B. Le composé GW se lie à SCAP, favorisant ainsi la translocation du complexe SCAP/SREBP même en présence d’un excès de cholestérol. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.