")
")
| Issue |
Med Sci (Paris)
Volume 42, Number 3, Mars 2026
|
|
|---|---|---|
| Page(s) | 234 - 237 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/2026027 | |
| Published online | 20 mars 2026 | |
Effet protecteur d’un variant « gain de fonction » de CREB3 dans la sclérose latérale amyotrophique
Protective effect of a CREB3 gain-offunction variant in amyotrophic lateral sclerosis
Université de Strasbourg, Inserm UMRS1329, Neurosciences et psychiatrie translationnelles de Strasbourg (STEP), Centre de recherche en biomédecine de Strasbourg (CRBS), Strasbourg, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
**
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
***
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
La sclérose latérale amyotrophique (maladie de Charcot) est une maladie neurodégénérative rare caractérisée par la perte des neurones moteurs supérieurs dans le cortex moteur, et des motoneurones dans le tronc cérébral et la moelle épinière [1]. Elle touche principalement les adultes autour de 65 ans en moyenne, se caractérise par des faiblesses et fasciculations musculaires accompagnées de dénervations et d’atrophie des muscles striés squelettiques, et progresse rapidement vers une paralysie [1]. Ces manifestations mènent généralement au décès par insuffisance respiratoire dans les 3 à 5 ans suivant l’apparition des premiers symptômes [1]. En France, la sclérose latérale amyotrophique affecte environ 1 700 nouveaux individus chaque année, et touche actuellement environ 8 000 personnes1.
La plupart des cas de sclérose latérale amyotrophique sont sporadiques, mais il existe environ 10 % de cas familiaux, pour lesquels la plupart des gènes responsables ont été identifiés : les plus fréquemment impliqués sont C9ORF72 (C9orf72-SMCR8 complex subunit), TARDBP (transactive response DNA-binding protein), SOD1 (superoxide dismutase 1) et FUS (FUS RNA-binding protein) [2]. Outre cette hétérogénéité génétique, la sclérose latérale amyotrophique se caractérise également par une grande hétérogénéité dans sa présentation clinique initiale, l’âge d’apparition des symptômes, la vitesse de progression, et la durée de la maladie. Ce second type d’hétérogénéité pourrait en partie résulter de composants génétiques influant directement sur les mécanismes moléculaires de la maladie, pour leur part très conservés et concernant la réparation de l’ADN, le métabolisme des ARN et des protéines, le trafic intracellulaire, et le fonctionnement des mitochondries[2].
En France, seul le riluzole (Rilutek®), un médicament anti-glutamatergique, est actuellement indiqué dans le traitement de la sclérose latérale amyotrophique. Il ralentit la progression de la maladie et retarde le décès des patients de quelques mois. L’identification de gènes impliqués dans les cas familiaux a permis le développement de thérapies géniques prometteuses (SOD1, FUS) ou, au contraire, qui se sont avérées toxiques (C9ORF72, Ataxin 2) [3]. Le Tofersen, un oligonucléotide « anti-sens » dirigé contre SOD1 vient d’obtenir une autorisation d’utilisation compassionnelle en France, et une autorisation de mise sur le marché aux États-Unis, ouvrant ainsi la voie à la thérapie génique pour la sclérose latérale amyotrophique. En raison de la grande hétérogénéité génétique de la maladie, il paraît logique d’opposer au développement de thérapies personnalisées, ciblées sur les multiples gènes responsables déjà identifiés dans les cas familiaux et qui concernent environ 7 % des patients, celui de thérapies ciblant les voies de signalisation communément altérées chez tous les patients, qu’il s’agisse de formes familiales ou sporadiques de la maladie. Il convient donc de compléter les analyses génétiques visant à identifier de nouveaux gènes impliqués dans la maladie par des analyses épigénétiques, transcriptomiques, ou protéomiques, visant à identifier de nouvelles voies de signalisation impliquées elles-aussi. Ces approches ne sont pas nouvelles, mais la plupart des analyses multi-omiques effectuées jusqu’à présent ont concerné la moelle épinière et les motoneurones, et peu ont concerné les neurones moteurs supérieurs et le cortex moteur. Pourtant, les résultats de récentes études génétiques indiquent que la sclérose latérale amyotrophique serait en premier lieu une maladie des neurones glutamatergiques [4], dont font partie les neurones moteurs supérieurs, contrairement aux motoneurones, qui sont eux cholinergiques. En combinant des données de RNAseq obtenues dans notre laboratoire à partir de neurones moteurs supérieurs purifiés provenant d’un modèle murin de sclérose latérale amyotrophique à des âges présymptomatiques et symptomatiques, avec des données de snR-NAseq (single nuclei RNA sequencing)2 obtenues post mortem à partir de tissus provenant de personnes atteintes de la maladie (cas sporadiques, ou cas familiaux par mutation du gène C9ORF72) [5], nous avons réalisé la première analyse transcriptomique interespèce pour cette maladie. Les premiers résultats de cette analyse ont été confrontés à des données génétiques et cliniques dans le but de découvrir les mécanismes moléculaires impliqués dans la progression de la maladie (Figure 1) et de proposer de nouvelles cibles thérapeutiques [6].
 |
Figure 1 Stratégie expérimentale ayant permis d’identifier le rôle neuroprotecteur d’un variant de CREB3 dans la sclérose latérale amyotrophique. A. L’intégration de données de séquençage des ARN messagers 1) de neurones moteurs supérieurs (NMS) purifiés à partir d’un modèle murin de sclérose latérale amyotrophique (SLA) (par la technique de RNAseq) et 2) contenus dans les noyaux cellulaires issus du cortex moteur de patients atteints de sclérose latérale amyotrophique (par la technique de snRNAseq) a permis l’identification des populations neuronales vulnérables à la maladie. B. Une analyse de coexpression des gènes dérégulés dans les populations neuronales d’intérêt (WGCNA) a mis en évidence un module (ensemble de gènes) surexprimé dans la maladie. Ce module correspond à une réponse neuronale au stress du réticulum endoplasmique, et constitue une signature de résilience orchestrée par le facteur de transcription CREB3. C. La combinaison des données de génétique humaine avec des données médicales a permis l’identification du rôle neuroprotecteur du variant rare CREB3R119G, qui protège les individus contre le risque de sclérose latérale amyotrophique, et ralentit la vitesse de progression de la maladie chez les personnes qui en sont atteintes (augmentation de la survie avec la maladie de 12 mois en moyenne). Figure réalisée avec BioRender. |
Analyse transcriptomique inter-espèces et identification d’une signature de résilience neuronale
Les neurones moteurs supérieurs constituent une population discrète du cortex moteur, et leur dégénérescence au cours de la sclérose latérale amyotrophique limite davantage leur présence dans les tissus humains post mortem. Pour pallier cette limitation et garantir la robustesse des résultats, nous avons combiné les données de RNAseq obtenues sur les neurones moteurs supérieurs murins avec celles de snRNAseq sur les tissus humains pour identifier et étudier un plus large éventail de neurones d’intérêt comprenant : 1) des neurones moteurs supérieurs de souris, 2) des neurones humains ayant la même identité cellulaire que les précédents (i.e., les neurones moteurs supérieurs humains), et 3) des neurones humains comportant les mêmes altérations transcriptionnelles chez les patients que les neurones moteurs supérieurs chez les souris modélisant la sclérose latérale amyotrophique (i.e., une population de neurones glutamatergiques des couches 2 et 3 du cortex moteur à projection intratélencéphalique) (Figure 1A) [6].
Pour identifier des voies de dérégulations robustes, nous avons soumis le groupe de neurones d’intérêt à une analyse pondérée des réseaux de coexpression de gènes (weighted co-expression gene network analysis, WGCNA), qui a notamment mis en évidence l’augmentation de l’expression d’un module de gènes impliqués dans la traduction des ARNm en protéines et le contrôle du stress du réticulum endoplasmique (Figure 1B), une voie de signalisation précédemment mise en cause dans la sclérose latérale amyotrophique [2]. Cependant, nous avons constaté que l’activation de cette voie de signalisation n’est pas restreinte aux neurones qui dégénèrent dans la maladie, mais qu’elle est commune à toutes les populations neuronales, ce qui suggère qu’elle constitue davantage une signature de résilience neuronale qu’une signature de neurodégénérescence, et ce qui modifie la manière d’envisager sa modulation dans une perspective thérapeutique [6].
Analyse génétique, données cliniques, et identification d’un variant rare du gène CREB3
En partant de l’hypothèse qu’une signature neuroprotectrice devrait être amplifiée chez les patients dont la maladie progresse lentement par rapport à ceux pour lesquels la progression est rapide, et que cette signature pourrait être influencée par les caractéristiques génétiques des individus, nous avons cherché à identifier un facteur de transcription qui régulerait conjointement l’expression des gènes du module de résilience, et présentant de potentiels variants rares dans les bases de données génétiques des personnes atteintes de la maladie (Figure 1B). Nous avons ainsi identifié le gène CREB3 (cAMP-responsive element-binding protein 3), dont le variant rare CREB3R119G est plus fréquent chez les individus sains que chez ceux atteints de sclérose latérale amyotrophique et diminue de 40 % le risque de développer la maladie. De plus, sa présence chez les patients freine significativement la progression de la maladie et augmente la survie de 12 mois en moyenne (Figure 1C). Ce variant CREB3R119G majore l’activité transcriptionnelle de la protéine CREB3, dont nous avons montré le caractère neuroprotecteur par la corrélation entre cette activité mesurée dans le cerveau post mortem ou dans le sang des patients et leur temps de survie avec la maladie [6].
CREB3 : une piste pour la thérapie génique de la sclérose latérale amyotrophique ?
CREB3 appartient à la famille des facteurs de transcription se liant aux éléments de réponse à l’AMP cyclique (cAMP-responsive elements, CRE) [6]. En réponse à un stress du réticulum endoplasmique, dans la membrane duquel est ancrée la protéine CREB3, elle est acheminée jusqu’à l’appareil de Golgi par des vésicules de transport COPII. Dans l’appareil de Golgi, elle est clivée par des protéases (S1P/S2P), ce qui permet le relargage de sa partie N-terminale. Ce fragment migre vers le noyau cellulaire, où il agit comme facteur de transcription sous forme d’homodimère ou d’hétérodimère en combinaison avec d’autres membres de la famille CREB (CREB3L1, CREB3L2) (Figure 2) [7]. CREB3 contrôle ainsi l’expression de gènes impliqués dans la réponse au stress du réticulum endoplasmique [7], ainsi que dans l’homéostasie calcique et mitochondriale [8]. Le rôle de CREB3 dans les neurones est encore mal connu, au-delà de son implication dans la régénération axonale de neurones sensoriels après une lésion chez le rat, notamment en induisant la synthèse de cholestérol [9]. La neuroprotection conférée par le variant CRE-B3R119G dans la sclérose latérale amyotrophique pourrait s’expliquer par une plus grande disponibilité de la forme clivée de CREB3 (plus grande sensibilité de la protéine au stress cellulaire, protéolyse activatrice de CREB3 plus efficace), ou par sa plus grande activité transcriptionnelle (plus grande affinité de liaison à l’ADN, capacité accrue à recruter la machinerie transcriptionnelle), autant de possibilités à explorer.
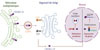 |
Figure 2 Mécanisme d’activation du facteur de transcription CREB3. La forme longue (CREB3-FL), non clivée, de la protéine CREB3 est ancrée dans la membrane du réticulum endoplasmique. En réponse à un stress du réticulum endoplasmique, la protéine est transportée vers l’appareil de Golgi, où elle est clivée par les protéases membranaires S1P et S2P. Le fragment clivé (CREB3-CF) rejoint le noyau cellulaire, où il peut être séquestré par un facteur de rétention spécifique (LRF) dans des corps nucléaires, ou bien se dimériser pour activer la transcription de gènes cibles. Figure réalisée avec BioRender. |
Parce que le facteur de transcription CREB3 est au cœur d’un mécanisme de résilience cellulaire, et que son degré d’activité est corrélé à la survie des personnes atteintes de sclérose latérale amyotrophique, quel que soit leur statut quant aux gènes responsables de la maladie, les variants « gain de fonction » de CREB3 sont des candidats prometteurs pour une thérapie génique dont pourraient bénéficier tous les patients afin de ralentir la progression de la maladie. Une telle thérapie génique, développée en parallèle de celles destinées à corriger spécifiquement le défaut génétique responsable de la maladie lorsque celui-ci a pu être identifié, a déjà été proposée dans d’autres maladies neurodégénératives, notamment la maladie de Parkinson [10], et permettrait d’apporter une réponse thérapeutique à la grande majorité des patients atteints de formes sporadiques ou familiales de sclérose latérale amyotrophique pour lesquelles le développement de thérapies ciblant la mutation responsable de la maladie s’avère difficile [3].
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article
Références
- van Es MA, Hardiman O, Chio A, Al-Chalabi A, et al. Amyotrophic lateral sclerosis. Lancet 2017 ; 390 : 2084–98. [Google Scholar]
- Goutman SA, Hardiman O, Al-Chalabi A, et al. Emerging insights into the complex genetics and pathophysiology of amyotrophic lateral sclerosis. Lancet Neurol 2022 ; 21 : 465–79. [Google Scholar]
- Ou K, Jia Q, Li D, et al. Application of antisense oligonucleotide drugs in amyotrophic lateral sclerosis and Huntington’s disease. Transl Neurodegener 2025 ; 14 : 4. [Google Scholar]
- van Rheenen W, van der Spek RAA, Bakker MK, et al. Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nat Genet 2021 ; 53 : 1636–48. [Google Scholar]
- Pineda SS, Lee H, Ulloa-Navas MJ, et al. Single-cell dissection of the human motor and prefrontal cortices in ALS and FTLD. Cell 2024 ; 187 : 1971–89. [Google Scholar]
- Megat S, Marques C, Hernán-Godoy M, et al. CREB3 gain-of-function variants protect against ALS. Nat Commun 2025 ; 16 : 2942. [Google Scholar]
- Sampieri L, Di Giusto, P, Alvarez C. CREB3 transcription factors: ER-Golgi stress transducers as hubs for cellular homeostasis. Front Cell Dev Biol 2019 ; 7 : 123. [Google Scholar]
- Smith BS, Hewitt T, Bakovic M, Lu R. ER stress-associated transcription factor CREB3 is essential for normal Ca2+, ATP, and ROS homeostasis. Mitochondrion 2023 ; 69 : 10–17. [Google Scholar]
- Ying Z, Zhai R, McLean NA, et al. The unfolded protein response and cholesterol biosynthesis link Luman/ CREB3 to regenerative axon growth in sensory neurons. J Neurosci 2015 ; 35 : 14557–70. [Google Scholar]
- Palfi S, Gurruchaga JM, Lepetit H, et al. Long-term follow-up of a phase I/II study of ProSavin, a lentiviral vector gene therapy for Parkinson’s disease. Hum Gene Ther Clin Dev 2018 ; 29 : 148–55. [Google Scholar]
Source : arsla.org
Cette technique permet d’analyser les produits de transcription des gènes des tissus difficiles à dissocier ou congelés (notamment des tissus post mortem) en isolant les noyaux cellulaires, ce qui évite les artefacts liés au stress de dissociation des cellules dans la technique scRNA-seq (single cell RNA sequencing) et offre une meilleure représentation de certains types cellulaires fragiles ou volumineux. En revanche, elle fournit un profil transcriptomique moins complet que la technique scRNAseq, puisqu’elle capte principalement les ARN pré-messagers (non encore épissés) qui sont présents dans le noyau, et ne permet pas d’accéder aux transcrits matures (épissés), qui sont dans le cytoplasme.
© 2026 médecine/sciences – Inserm
 Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Liste des figures
|
Figure 1 Stratégie expérimentale ayant permis d’identifier le rôle neuroprotecteur d’un variant de CREB3 dans la sclérose latérale amyotrophique. A. L’intégration de données de séquençage des ARN messagers 1) de neurones moteurs supérieurs (NMS) purifiés à partir d’un modèle murin de sclérose latérale amyotrophique (SLA) (par la technique de RNAseq) et 2) contenus dans les noyaux cellulaires issus du cortex moteur de patients atteints de sclérose latérale amyotrophique (par la technique de snRNAseq) a permis l’identification des populations neuronales vulnérables à la maladie. B. Une analyse de coexpression des gènes dérégulés dans les populations neuronales d’intérêt (WGCNA) a mis en évidence un module (ensemble de gènes) surexprimé dans la maladie. Ce module correspond à une réponse neuronale au stress du réticulum endoplasmique, et constitue une signature de résilience orchestrée par le facteur de transcription CREB3. C. La combinaison des données de génétique humaine avec des données médicales a permis l’identification du rôle neuroprotecteur du variant rare CREB3R119G, qui protège les individus contre le risque de sclérose latérale amyotrophique, et ralentit la vitesse de progression de la maladie chez les personnes qui en sont atteintes (augmentation de la survie avec la maladie de 12 mois en moyenne). Figure réalisée avec BioRender. |
| Dans le texte | |
|
Figure 2 Mécanisme d’activation du facteur de transcription CREB3. La forme longue (CREB3-FL), non clivée, de la protéine CREB3 est ancrée dans la membrane du réticulum endoplasmique. En réponse à un stress du réticulum endoplasmique, la protéine est transportée vers l’appareil de Golgi, où elle est clivée par les protéases membranaires S1P et S2P. Le fragment clivé (CREB3-CF) rejoint le noyau cellulaire, où il peut être séquestré par un facteur de rétention spécifique (LRF) dans des corps nucléaires, ou bien se dimériser pour activer la transcription de gènes cibles. Figure réalisée avec BioRender. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.