")
")
| Issue |
Med Sci (Paris)
Volume 37, Novembre 2021
Les Cahiers de Myologie
|
|
|---|---|---|
| Page(s) | 32 - 35 | |
| Section | Prise en charge | |
| DOI | https://doi.org/10.1051/medsci/2021190 | |
| Published online | 8 décembre 2021 | |
Le suivi multidisciplinaire de patients adultes atteints de dystrophie myotonique de type 1 dans le Sud Aquitain
Multidisciplinary care of patients with Myotonic Dystrophy type 1 (DM1) in South Aquitaine
1
Centre de Compétence des Maladies Neuromusculaires de Bayonne, France
2
Centre de Compétence des Maladies Neuromusculaires de Hendaye, France
Abstract
DM1 is characterized by a multisystemic involvement. Our objective was to determine the proportion of adequate follow-up for each affected organ in DM1 patients based on the recently published American and Spanish recommendations. To this end, we conducted a descriptive cross-sectional survey by phone in adult, genetically proven DM1 patients followed in the two French neuromuscular centers of Bayonne and Hendaye located in South Aquitaine, France. The questionnaire selected the most stringent criteria of the two international recommendations for each item of follow-up. Seventy-three patients were included, 55% of which were women (mean age of 48 years) with an average number of 467 CTG repeats. The proportion of patients receiving clinical follow-up in accordance with the recommendations was 90% in cardiology, 60% in neurology, 68% in ophthalmology, 53% in physiotherapy, 23% in pneumology, and 12% in rehabilitation. The high rate of neurological, cardiological, and ophthalmological monitoring might be explained by a locally dense medical demography whereas low rate of respiratory follow up and rehabilitation may reflect an incomplete knowledge of both the disease and the questionnaire. These results should be carefully interpretated as cognitive status may influence such a declarative study. Our study nevertheless disclosed important disparities according to the recommended multidisciplinary follow-up criteria in this French cohort of adult DM1 patients. These results highlight the major role of a multidisciplinary care and monitoring in DM1.
© 2021 médecine/sciences – Inserm

© Valérie Allamand/Servier Medical Art
Introduction
La dystrophie myotonique de type 1 (DM1) ou maladie de Steinert représente une des formes les plus fréquentes de myopathie chez l’adulte. Outre un déficit musculaire potentiellement invalidant et nécessitant un suivi rééducatif et neurologique régulier, la DM1 se caractérise aussi par une atteinte multisystémique nécessitant des évaluations régulières par différents spécialistes d’organes tels que les cardiologues, pneumologues, ophtalmologues, endocrinologues, hépato-gastro-entérologues, et neuropsychologues. En 2018, un consortium d’experts internationaux mandatés par la Myotonic Dystrophy Association a publié des recommandations pour le diagnostic et la prise en charge de la DM1 [1]. L’année suivante, un groupe d’experts faisait de même pour l’Espagne [2]. Ces recommandations soulignaient la nécessité d’une organisation structurée et adaptée dans le suivi multidisciplinaire. La densité de patients DM1 est particulièrement élevée dans le Sud Aquitain du fait de la proximité avec le Pays Basque espagnol. La prévalence de la maladie y est en effet de 36/100 000 en Navarre [3] et de 26,5/100 000 dans la province basque du Guipúzcoa [4].
Objectif, matériels et méthodes
L’objectif de notre étude était de déterminer la proportion de patients adultes atteints de DM1 et suivis dans les deux Centres de Compétence neuromusculaire du Sud Aquitain (CCNM), bénéficiant d’un suivi médical en adéquation avec ces recommandations internationales. À cette fin, nous avons réalisé une enquête descriptive transversale par questionnaire téléphonique auprès de patients DM1 adultes et de leur aidant principal. Les patients atteints de DM1 représentent de loin la population la plus importante de patients neuromusculaires suivis dans les deux CCNM. L’étude a fait l’objet d’une déclaration de conformité de type MR4 auprès de la CNIL (référence n° 2214338). La population source était représentée par l’ensemble des patients adultes atteint de DM1 ayant déjà été vus au moins une fois en consultation neuromusculaire entre janvier 1998 et mai 2019 dans l’un des deux CCMN des Pyrénées-Atlantiques (l’hôpital Marin de Hendaye – appartenant à l’APHP – et le Centre Hospitalier de la Côte Basque à Bayonne). Les consultations multidisciplinaires sont assurées dans les deux centres en fonction du lieu d’habitation du patient, et en parfaite coordination. Les critères d’inclusion étaient : patient adulte atteint d’une DM1 génétiquement prouvée (nombre de répétitions CTG ⩾ 50), ayant donné son consentement pour participer à l’étude.
Du fait de différences entre les deux recommandations internationales prises comme référentiels, le questionnaire a été établi en privilégiant les exigences de suivi les plus rigoureuses en termes d’exhaustivité et de fréquence de suivi pour ce qui concerne les évaluations cliniques et paracliniques (Tableau I).
Périodicité des évaluations cliniques spécialisées et des examens paracliniques choisie selon les recommandations les plus rigoureuses américaines (US) [1] ou espagnoles (ES) [2] pour la DM1.
Résultats
Soixante-treize patients ont pu être inclus, soit un taux de participation de 95 % dont 55 % de femmes ( Figure 1 ). L’âge moyen était de 48 ans et le nombre de répétition CTG était en moyenne de 467. Les proportions de patients bénéficiant d’un suivi clinique et paraclinique conforme aux recommandations sont détaillées dans la Figure 2 . On observe que plus de la moitié des patients bénéficient d’un suivi conforme pour ce qui concerne le suivi cardiologique (90 %), le suivi ophtalmologique (68 %), le suivi neurologique et odontologique (60 %). En revanche, peu de patients bénéficient d’un suivi conforme aux recommandations dans le domaine rééducatif (< 18 %) ou en pneumologie (23 %).
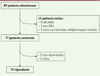 |
Figure 1. Design de l’étude. |
 |
Figure 2. Résultats de l’enquête téléphonique concernant le suivi conforme aux recommandations. |
Discussion
Dans cette enquête visant à évaluer l’exhaustivité du suivi des patients DM1 en regard des dernières recommandations internationales, nous observons que 60 % des patients avaient un suivi neurologique conforme en ayant consulté un neurologue au cours de l’année écoulée. Ce chiffre est similaire à celui retrouvé dans une enquête de l’AFM-Téléthon (55 %) réalisée en 2011 [5], et à celui de l’étude américaine « The Christopher Project » (49 %) datant de 2019 [6]. Ce suivi neurologique apparemment imparfait en CCMN, pourrait être expliqué par une observance parfois faible de patients présentant des difficultés cognitives, ou par une adaptation personnalisée du suivi selon la sévérité de l’atteinte, ce qui ne figure pas dans les recommandations.
En fonction de l’organe à surveiller, on constate une hétérogénéité de suivi. Dans notre cohorte, 90 % du suivi cardiologique est assuré conformément aux recommandations, à savoir annuellement, ce qui représente un chiffre supérieur à ceux rapportés précédemment de 44 % [5] et 74 % [6]. L’importance du suivi cardiologique pourrait être expliqué par l’implication thérapeutique engendrée par le dépistage des troubles conductifs et rythmiques, puisqu’un tiers des décès sont d’origine cardio-vasculaire [7, 8] dans cette pathologie, mais aussi par la démographie médicale dense localement permettant un accès facilité aux soins.
À l’inverse, le suivi pneumologique avec réalisation d’une spirométrie tous les six mois ne concerne que 23 % et 16 % des patients, alors que l’atteinte respiratoire constitue la principale cause de morbi-mortalité chez les patients DM1 en général [9, 10]. Il n’existe pas de données comparatives, mais ces chiffres bas ne semblent pas refléter un défaut de connaissance de cette atteinte dans la DM1, puisque l’accès à une évaluation initiale par un pneumologue est supérieur à 65 % chez les patients interrogés. Ils mettent donc plus en lumière un défaut de suivi dans le temps d’une éventuelle atteinte pulmonaire. Il faut noter que les recommandations publiées ne permettent pas un suivi personnalisé pour les patients DM1 qui seraient plus à risque d’atteinte respiratoire, et qui nécessiteraient par conséquent une fréquence de suivi plus importante selon certains facteurs de risque : par exemple, la taille de l’expansion CTG, puisqu’il existe une corrélation avec l’intensité de l’atteinte respiratoire [11, 12], mais également le sexe masculin, l’obésité [13, 14], ou encore l’âge, puisque cette atteinte survient classiquement entre 50 et 60 ans [15]. De même, les patients atteints de forme tardive n’ont pas nécessairement besoin d’une évaluation respiratoire bi-annuelle.
Le défaut de suivi est aussi marqué sur le plan rééducatif, puisque seuls 12 % des patients interrogés étaient suivis annuellement par un médecin de Médecine Physique et de Réadaptation (MPR), 18 % avaient un suivi ergothérapique annuel, et 12 % bénéficiaient d’un suivi orthophonique au moins annuel. Ces résultats sont comparables à ceux de l’étude intitulée The Christopher Project [6]. En revanche, 53 % bénéficiaient de séances de kinésithérapie au moins hebdomadaires. Ces résultats semblent insuffisants en comparaison de ceux publiés en 1999 par une équipe de rééducateurs retrouvant un suivi régulier par un médecin rééducateur chez 48 % de patients présentant différents types de maladies neuromusculaires, cette étude ayant néanmoins ciblé plusieurs pathologies neuromusculaires [16]. L’accès à ces professionnels de santé, notamment les médecins MPR, est parfois compliqué, et nécessite une organisation en réseau du fait de leur faible densité au niveau national et de leur mode d’exercice majoritairement hospitalier. D’autre part, le suivi par un ergothérapeute n’est pas nécessairement adapté à tous les patients, et devrait faire l’objet de recommandations personnalisées. Enfin, signalons qu’il s’agit d’une étude déclarative concernant parfois des patients avec atteinte cognitive et que certaines terminologies ont pu être imparfaitement comprises, en particulier celles de médecin rééducateur ou d’ergothérapeute.
Conclusion
Il existe d’importantes disparités dans l’exhaustivité du suivi multidisciplinaire de la DM1 au sein de la cohorte de patients interrogés. Les résultats sont toutefois à prendre avec précaution s’agissant d’une étude déclarative, et concernent des recommandations qui ne tiennent pas compte de la sévérité individuelle du patient. Celles-ci devraient normalement imposer un suivi personnalisé selon la gravité de l’atteinte d’organe. Cette étude met en lumière le rôle majeur de la coordination du suivi multidisciplinaire, et pourrait amener à modifier les pratiques.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Remerciements
Nous remercions J. Andoni Urtizberea et Ana-Maria Cobo (précédemment en charge du Centre de compétence neuromusculaire de l’Hôpital Marin de Hendaye) pour leur contribution à ce travail.
Ce travail a fait d’une thèse de doctorat en médecine générale soutenue par Thomas Bisson le 8 avril 2021 à Bordeaux et ayant obtenu la mention très honorable.
Références
- Ashizawa T, Gagnon C, Groh WJ, et al. Consensus-based care recommendations for adults with myotonic dystrophy type 1. Neurol Clin Pract 2018 ; 8 : 507–520. [CrossRef] [PubMed] [Google Scholar]
- Gutiérrez Gutiérrez G, Díaz-Manera J, Almendrote M, et al. Clinical guide for the diagnosis and follow-up of myotonic dystrophy type 1, MD1 or Steinert’s disease. Med Clin (Barc) 2019; 153 : 82.e1-17. [CrossRef] [Google Scholar]
- Pagola-Lorz I, Vicente E, Ibáñez B, et al. Epidemiological study and genetic characterization of inherited muscle diseases in a northern Spanish region. Orphanet J Rare Dis 2019 ; 14 : 276. [CrossRef] [PubMed] [Google Scholar]
- López de Munain A, Blanco A, Emparanza JI, et al. Prevalence of myotonic dystrophy in Guipúzcoa (Basque country, Spain). Neurology 1993 ; 43 : 1573–1576. [CrossRef] [PubMed] [Google Scholar]
- AFM. Enquête auprès de personnes atteintes de la dystrophie myotonique de Steinert. AFM-Téléthon, Myobase, 2011. [Google Scholar]
- Howe SJ, Marigold foundation, The Christopher project reference group. The Christopher Project. Report to the myotonic dystrophy community. Myotonic 2019. https://www.myotonic.org/sites/default/files/pages/files/Christopher_Project_Full_Report.pdf. [Google Scholar]
- Merlevede K, Vermander D, Theys P, et al. Cardiac involvement and CTG expansion in myotonic dystrophy. J Neurol 2002 ; 249 : 693–698. [CrossRef] [PubMed] [Google Scholar]
- Groh WJ, Groh MR, Saha C, et al. Electrocardiographic abnormalities and sudden death in myotonic dystrophy type 1. N Engl J Med 2008 ; 358 : 2688–2697. [CrossRef] [PubMed] [Google Scholar]
- de Die-Smulders CE, Höweler CJ, Thijs C, et al. Age and causes of death in adult-onset myotonic dystrophy. Brain 1998 ; 121 : 1557–1563. [CrossRef] [PubMed] [Google Scholar]
- Mathieu J, Allard P, Potvin L, et al. A 10-year study of mortality in a cohort of patients with myotonic dystrophy. Neurology 1999 ; 52 : 1658–1662. [CrossRef] [PubMed] [Google Scholar]
- Monteiro R, Bento J, Gonçalves MR, et al. Genetics correlates with lung function and nocturnal ventilation in myotonic dystrophy. Sleep Breath 2013 ; 17 : 1087–1092. [CrossRef] [PubMed] [Google Scholar]
- Boussaïd G, Wahbi K, Laforet P, et al. Genotype and other determinants of respiratory function in myotonic dystrophy type 1. Neuromuscul Disord 2018 ; 28 : 222–228. [CrossRef] [PubMed] [Google Scholar]
- Dogan C, De Antonio M, Hamroun D, et al. Gender as a modifying factor influencing myotonic dystrophy type 1 phenotype severity and mortality: a nationwide multiple databases cross-sectional observational study. PLoS One 2016 ; 11 : e0148264. [CrossRef] [PubMed] [Google Scholar]
- Seijger CGW, Drost G, Posma JM, et al. Overweight is an independent risk factor for reduced lung volumes in myotonic dystrophy type 1. PLoS One 2016 ; 11 : e0152344. [CrossRef] [PubMed] [Google Scholar]
- Groh WJ, Groh MR, Shen C, et al. Survival and CTG repeat expansion in adults with myotonic dystrophy type 1. Muscle Nerve 2011 ; 43 : 648–651. [CrossRef] [PubMed] [Google Scholar]
- Donzé C, Delattre S, Viet G, et al. Neuromuscular disease: health care accessibility in the Nord-Pas-de-Calais region. Rev Neurol (Paris) 1999 ; 155 : 1063–1070. [PubMed] [Google Scholar]
Liste des tableaux
Périodicité des évaluations cliniques spécialisées et des examens paracliniques choisie selon les recommandations les plus rigoureuses américaines (US) [1] ou espagnoles (ES) [2] pour la DM1.
Liste des figures
|
Figure 1. Design de l’étude. |
| Dans le texte | |
|
Figure 2. Résultats de l’enquête téléphonique concernant le suivi conforme aux recommandations. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.