")
")
| Issue |
Med Sci (Paris)
Volume 17, Number 12, Décembre 2001
|
|
|---|---|---|
| Page(s) | 1281 - 1288 | |
| Section | Articles de Synthèse | |
| DOI | https://doi.org/10.1051/medsci/200117121281 | |
| Published online | 15 December 2001 | |
TWIST : un nouvel acteur de l’ossification des os plats
TWIST: a new piece to the puzzle on flat bones ossification
Inserm U. 393, Hôpital Necker-Enfants Malades, 149, rue de Sèvres, 75743 Paris Cedex 15, France
Résumé
Le syndrome de Saethre-Chotzen est une malformation osseuse qui résulte de la fusion prématurée de certaines sutures de la voûte du crâne (craniosténose), associée à des anomalies du visage et des extrémités des membres. L’identification de mutations du gène TWIST chez les patients atteints du syndrome de Saethre-Chotzen a propulsé, sur la scène de l’ostéoblaste, ce petit facteur de transcription à domaine basique hélice-boucle-hélice d’abord connu pour son rôle précoce dans l’induction ou la spécification du mésoderme. Toutes les mutations identifiées chez les patients affectent le domaine bHLH de TWIST et induisent, par des mécanismes distincts, une perte de fonction de la protéine. Des données récentes indiquent que TWIST exercerait un rôle inhibiteur sur la différenciation ostéoblastique associée à une modulation de l’expression des FGFR, gènes dont des mutations sont également responsables de craniosténoses syndromiques. De nombreuses autres malformations de la voûte crânienne ont révélé l’implication dans l’ostéogenèse des os plats, d’autres facteurs de transcription comme MSX2, ALX4 et CBFA1/RunX2. Les efforts se concentrent désormais sur la compréhension des relations fonctionnelles susceptibles d’intervenir entre ces différents protagonistes de l’ossification membranaire.
Abstract
TWIST is a highly conserved developmental gene that encodes a basic helix-loop-helix transcription factor involved in early mesoderm patterning. Few years ago, identification of TWIST loss-of-function mutations in the Saethre-Chotzen syndrome (SCS), a skull disorder called craniosynostosis and associated with facial and limbs anomalies, has revealed an unexpected implication of this factor in flat bones osteogenesis. Recent data from both mouse and human indicate that TWIST can modulate expression of fibroblast growth factor receptors (FGFRs), a family of genes involved in conditions clinically close to SCS. Although TWIST direct target genes remain to be identified in osteoblasts, several lines of evidence suggest that TWIST could prevent premature ossification of skull sutures by negatively regulating osteoblastic differentiation. Both cellular and mouse models will be helpful to determine the precise role of this transcription factor in flat bone development and its functional relationships with other protagonists of skull sutures differentiation.
© 2001 médecine/sciences - Inserm / SRMS
Contrairement à celui des os longs, le processus d’ossification des os plats (ou os de membrane) implique la différenciation directe de cellules du mésenchyme et de la crête neurale en ostéoblastes sans l’intervention d’une structure cartilagineuse intermédiaire. Il en va ainsi de l’ostéogenèse de la voûte du crâne qui se constitue tardivement au cours de la période fœtale par expansion osseuse progressive etconcentrique à partir de plusieurs centres d’ossification membranaire. Ces différentes pièces osseuses se développent de façon apparemment indépendante à la surface de l’encéphale où elles finissent par se rejoindre au niveau de sutures et de fontanelles, structures dynamiques dont la persistance après la naissance assure une souplesse et une adaptabilité du crâne indispensables au développement harmonieux du cerveau (figure 1).
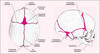 |
Figure 1. Représentation schématique du crâne d’un nouveau-né. Les différents os qui composent la voûte crânienne se rencontrent à la naissance sans pour autant fusionner complètement: les frontières entre deux os délimitent une suture alors que les carrefours entre plus de deux os forment une fontanelle (adapté d’après [30]). |
Les craniosténoses sont des anomalies très fréquentes de l’ossification du crâne (1/3 000 naissances) qui résultent de la fusion prématurée d’une ou de plusieurs sutures. Elles induisent des déformations de la boîte crânienne plus ou moins marquées selon la nature et le nombre de sutures synostosées, et peuvent engendrer, dans les cas les plus sévères, des cécités par compression des nerfs optiques et des retards mentaux par compression cérébrale. Plus rarement, elles sont associées à des anomalies de la face et des extrémités des membres:on parle alors de craniosténoses syndromiques. On sait depuis 1995 que la majorité des craniosténoses syndromiques transmises sur le mode autosomique dominant (notamment les syndromes d’Apert, de Pfeiffer et de Crouzon) sont liées à des mutations activatrices des gènes FGFR (fibroblast growth factor receptor) qui codent pour les récepteurs des facteurs de croissance fibroblastiques (m/s 1994, n°11, p. 1163 et 1995, n° 12, p. 1748) (Tableau I). Plus récemment, nous avons montré, en même temps qu’un groupe américain, l’implication du gène Twist dans une autre craniosténose autosomique dominante, le syndrome de Saethre-Chotzen [1, 2] (m/s 1997, n° 4, p. 576). Ce syndrome se caractérise par une fusion prématurée des sutures coronales associée à une dysmorphie faciale discrète mais très caractéristique (notamment une implantation basse des cheveux, des oreilles petites et rondes et un ptôsis, c’est-à-dire une paupière tombante). Une syndactylie cutanée, exclusivement entre l’index et le majeur, et un élargissement du gros orteil sont aussi très fréquemment observés dans cette maladie. Twist est quant à lui un gène d’expression embryonnaire précoce identifié initialement chez la drosophile chez laquelle son absence totale entraîne l’échec de la gastrulation et la mort de l’embryon avec un phénotype «vrillé» (twisted). L’identification de ce gène de développement dans une maladie de l’ossification membranaire nous a dès lors conduits à formuler l’hypothèse d’un rôle de Twist, chez les vertébrés, dans le contrôle de l’ostéogenèse des os plats.
Les principales craniosténoses syndromiques transmises sur le mode autosomique dominant : dentification des gènes et des mécanismes impliqués.
Différents mécanismes de perte de fonction de Twist sont responsables du syndrome de Saethre-Chotzen
Le gène Twist comprend deux exons séparés par un intron, mais la séquence codante (609 paires de bases chez l’homme) est entièrement incluse dans le premier exon et code pour une protéine de la famille des facteurs de transcription à domaine bHLH (basic helix-loop-helix). Ces facteurs, maintenant bien connus, se lient à l’ADN sous forme de dimères et sont impliqués dans la modulation précoce de nombreux processus développementaux comme la neurogenèse, la myogenèse ou plus généralement la spécification de différents lignages cellulaires. Plus de 250 facteurs de cette grande famille ont déjà été décrits et leur classification est fondée sur leur spécificité d’expression ou sur la structure du motif bHLH en association ou non avec d’autres types de domaines protéiques [3]. Ainsi, Twist est classique ment reconnu comme un facteur bHLH de classe B, c’est-à-dire dont l’expression est spécifique du tissu. Pendant l’embryogenèse, il s’exprime en effet essentiellement dans le feuillet mésodermique et les cellules de la crête neurale, puis plus spécifiquement encore, dans certains tissus qui en dérivent comme le mésenchyme céphalique, l’ébauche de la mandibule et des bourgeons de membres. Chez la souris (dont la structure du gène Twist est étonnamment conservée par rapport à l’homme, 100% d’identité dans le domaine bHLH), l’invalidation du gène donne un phénotype létal embryonnaire à l’état homozygote (à 11,5 jours de développement avec une sévère exencéphalie), et un phénotype osseux à l’état hétérozygote. Celui-ci mime les symptômes majeurs et même la variabilité clinique décrits dans le syndrome de Saethre-Chotzen [1, 4]. C’est d’ailleurs ce modèle hétérozygote qui, conjointement à la localisation du gène Twist humain sur le bras court du chromosome 7, a permis son identification en 1997 comme gène responsable de cette maladie [1, 2].
Plus de 50 mutations intragéniques hétérozygotes, touchant toutes le domaine bHLH de Twist, ont déjà été décrites chez des patients présentant ce syndrome (figure 2). Parmi elles, on trouve des mutations non sens, faux sens, des insertions/duplications avec ou sans décalage du cadre de lecture, et des délétions entraînant l’apparition prématurée d’un codon de terminaison de la traduction (figure 2A) [5, 6] (m/s 1999, n° 5, p. 761). Malgré l’abondance des types de mutations identifiés chez les différents patients, le syndrome de Saethre-Chotzen semblait, comme chez la souris, résulter d’une haplo-insuffisance au locus Twist, d’autant que des délétions hétérozygotes de la totalité du gène chez quelques patients venaient d’être décrites [7]. L’absence de corrélation entre la nature de la mutation et la sévérité clinique du phénotype (pourtant très variable d’un individu à l’autre) plaidait en outre en faveur d’une perte de fonction du produit de l’allèle muté, quel que soit le type de mutation. Plusieurs approches fonctionnelles ont récemment permis de le démontrer en mettant en lumière, à défaut d’une corrélation phénotype-génotype, une nette corrélation entre la nature de la mutation, sa position dans le domaine bHLH et le type de mécanisme engendrant une perte de la fonction de Twist [8, 9] (m/s 2000, n°8-9, p. 983). Ainsi, un premier niveau d’haplo-insuffisance intéresse les mutations non-sens qui n’empêchent pas la synthèse des protéines tronquées correspondantes mais induisent, dans des cellules Cos transfectées, leur dégradation en quelques heures. Les autres mutations ne semblent pas affecter la stabilité de la protéine. En revanche, lorsqu’une mutation faux sens touche l’une des deux hélices du domaine bHLH, la protéine Twist n’est pratiquement plus capable de former un dimère avec E12, facteur de la même famille issu du gène E2A, et d’expression ubiquitaire (bHLH de classe A). Ce second mécanisme de perte de fonction concorde avec l’observation que la protéine Twist doit interagir avec son partenaire pour pouvoir garder sa localisation nucléaire puisque les protéines dont les hélices a sont mutées ponctuellement montrent une localisation très majoritairement cytoplasmique. Un troisième mécanisme concerne les mutations du domaine basique du bHLH (domaine de liaison à l’ADN) et des résidus qui se trouvent à la jonction entre la boucle et la seconde hélice a. Dans ce cas, la protéine mutante est toujours capable de former un dimère avec E12 mais le complexe n’est plus capable de se fixer à l’ADN. De plus, les mutations qui affectent la boucle entre les deux hélices a (essentiellement des duplications en phase) diminuent mais n’empêchent com-plètement l’interaction ni le partenaire protéique, ni avec la cible nucléotidique. Enfin, bien qu’aucune instabilité des transcrits de Twist n’ait été mise en évidence à partir des cellules transfectées, un niveau d’haploinsuffisance supplémentaire pourrait néanmoins se manifester au stade de l’ARN messager porteur d’un codon stop, puisque des ostéoblastes immortalisés d’un patient hétérozygote pour la mutation Y103X (qui tronque toute la partie bHLH de Twist) ont montré une réduction du transcrit par rapport aux ostéoblastes témoins [10]. Ce mécanisme de dégradation partielle ou totale des ARNm porteurs d’un codon de terminaison, appelé NMD (nonsense mediated mRNA decay), est maintenant clairement reconnu [11]. Il pourrait être associé, dans le cas de Twist, à un rôle du deuxième exon, transcrit mais non traduit (et absent des constructions utilisées avec les cellules Cos), dans la stabilité et l’intégrité du messager. Ces différents niveaux d’haplo-insuffisance sont récapitulés dans la figure 2B. En affectant la stabilité de Twist, son hétérodimérisation avec E12, sa localisation intracellulaire ou encore son affinité pour l’ADN cible, toutes les mutations identifiées chez les patients conduisent donc finalement à une perte de la fonction de Twist. Cependant, l’élucidation de la véritable fonction physiologique de ce facteur transcriptionnel dans la différenciation de l’os de membrane suppose, en premier lieu, l’identification des protéines avec lesquelles il interagit in vivo ainsi que des gènes qui sont soumis à son contrôle dans le contexte ostéoblastique.
  |
Figure 2. Le syndrome de Saethre-Chotzen résulte d’une haplo-insuffisance de Twist. A. Position des différentes mutations. Une cinquantaine de mutations du gène Twist (substitutions, mutations stop, délétions, insertions) ont été identifiées à l’etat hétérozygote chez de nombreux patients. Les mutations mentionnées en rouge prédisent la synthèse d’une protéine tronquée. B. Les différents niveaux d’haplo-insuffisance. L ’haploinsuffisance de Twist peut résulter de mécanismes distincts qui entrent en jeu à des étapes différentes de la synthèse ou de la fonction du facteur de transcription. Dans tous les cas, la mutation conduit finalement à une perte de fonction de Twist. |
Twist règle-t-il l’expression des gènes FGFR ?
Parmi les gènes susceptibles d’être réglés par Twist, ceux qui codent pour les récepteurs des facteurs de croissance fibroblastiques (FGFR) s’avéraient de séduisants candidats. Comme nous l’avons évoqué dans l’introduction, la majorité des craniosténoses syndromiques sont en effet liées à des mutations des trois premiers membres de cette famille de récepteurs à domaines tyrosine kinase, FGFR2 étant impliqué nettement plus fréquemment que FGFR1 et FGFR3 (Tableau I). Rappelons au passage que le quatrième membre de cette famille, FGFR4, n’est à ce jour associé à aucune maladie connue. Les syndromes les plus sévères présentent une symptomatologie qui permet un diagnostic relativement aisé (comme le syndrome d’Apert dans lequel une importante brachycéphalie est systématiquement associée à de graves fusions osseuses et cutanées des doigts et des orteils). On identifie alors géné-ralement la mutation causale dans le gène suspecté, en l’occurrence FGFR2. Mais dans de bien nombreux cas, la classification clinique est loin d’être évidente car la grande variabilité phénotypique (en particulier la variabilité intra-familiale) rend le diagnostic délicat. Ces ressemblances phénotypiques entre les craniosténoses liées aux FGFR et à Twist laissaient supposer une relation étroite entre ces acteurs moléculaires. En outre, l’expression des gènes FGFR1-3 et de Twist au niveau du crâne en particulier au niveau des différentes sutures, ne fait plus aucun doute et leur patron d’expression spatio-temporel a même été étudié avec précision chez la souris et l’homme [12–16]. Ces récentes études montrent que les trois FGFR et Twist sont exprimés au niveau des sutures en cours de formation. Cependant, l’expression de Twist précède de quelques jours celle des FGFR et concerne spécifiquement le mésenchyme non différencié qui sépare les deux fronts de la suture. Au contraire, FGFR1 est très exprimé dans les cellules ostéoblastiques mûres qui constituent la partie déjà ossifiée de l’os de membrane, alors que FGFR2 (et à un niveau moindre, FGFR3) sont retrouvés au niveau des fronts ostéogéniques qui affleurent le mésenchyme, c’est-à-dire dans les cellules ostéoblastiques prolifératives les plus immatures (figure3). Ces profils d’expression suggé-raient donc que Twist pouvait se situer en amont des FGFR, hiérarchie déjà établie chez la drosophile chez laquelle l’absence de Twist supprime ou perturbe l’expression des homologues des FGFR, DFR1 et DFR2 [17]. Dans la mesure où, chez les vertébrés, la craniosténose résulte soit d’une haplo-insuffisance de Twist (perte de fonction) soit d’une activation des FGFR (gain de fonction), l’hypothèse la plus vraisemblable était donc que Twist exerce un rôle inhibiteur, notamment sur FGFR2 dont l’expression dans les ostéoblastes est plus précoce que celle de FGFR1. De récentes analyses de l’expression de ces gènes, par hybridation in toto sur les embryons de souris hétérozygotes pour l’invalidation de Twist, semblent montrer qu’en effet, la diminution de l’expression de Twist est associée à une diminution de l’expression de FGFR1 et à une augmentation de celle de FGFR2 [18]. Ce résultat concorde avec la récente observation, par immunohistochimie sur des coupes des mêmes souriceaux hétérozygotes, d’une expression ectopique du récepteur FGFR2 au niveau du mésenchyme sutural, précisément là où l’expression de Twist est diminuée de moitié [14]. L’observation d’une surexpression de FGFR2 dans les ostéoblastes immortalisés d’un patient porteur d’une mutation tronquante de Twist vient également conforter cette hypothèse. Bien que la dynamique de fermeture des sutures et l’hétérogénéité évidente dans les différents stades de fusion demeure des obstacles majeurs à l’observation de différences d’expression constantes, le lien génétique entre Twist et FGFR2 (au moins) apparaît désormais évident. Dans la mesure où l’expression de FGFR2 est clairement associée à la zone proliférative des fronts ostéogéniques en regard du mésenchyme sutural, on pense aujourd’hui que Twist pourrait inhiber la prolifération des cellules pré-ostéoblastiques, en maintenant une faible expression de FGFR2 au niveau des fronts osseux bordant la suture (figure 3).
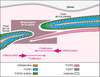 |
Figure 3. Expression des gènes FGFR1, FGFR2, FGFR3 et Twist au niveau de la suture coronale chez la souris à 16 jours de développement. Twist est exprimé dans le tissu mésenchymateux qui constitue la suture, entre les fronts ostéogéniques. Ces derniers sont le siège d’une prolifération intense des pré-ostéoblastes qui est corrélee à une forte expression de FGFR2. Cette dernière, plus élevée encore dans le contexte d’une haplo-insuffisance de Twist, précède l’etape de différenciation osseuse au cours de laquelle FGFR1 devient plus exprimé que FGFR2 (Modifié d’après [13]). |
Il reste néanmoins à déterminer si l’effet modulateur que Twist semble exercer sur les FGFR s’explique, au niveau moléculaire, par un contrôle direct ou indirect. Chez la souris comme chez l’homme, Twist est capable de former un hétérodimère stable avec E12 et de se lier sous cette forme à l’ADN au niveau de motifs hexanucléotidiques appelés boîtes E. Le complexe Twist/E12 reconnaît spécifiquement et avec une grande affinité la boîte CATATG, encore appelée boîte NdeI [9, 19]. L’étude de 3,5 kilobases du promoteur humain de FGFR2 n’a révélé la présence d’aucune boîte NdeI et malgré l’existence de plusieurs autres motifs CANNTG, aucun essai de validation fonctionnelle n’a encore permis de tester si l’effet modulateur que Twist semble exercer sur l’expression des gènes FGFR est direct ou indirect.
Twist:un modulateur négatif de la différenciation ostéoblastique
Avant l’identification de mutations de Twist dans le syndrome de Saethre-Chotzen, personne ne suspectait que ce facteur de transcription exprimé très tôt au cours du développement embryonnaire puisse jouer un quelconque rôle dans la différenciation ostéoblastique. Chez les vertébrés, Twist est progressivement exclu du myotome à mesure que le mésoderme se différencie, et de nombreuses études ont montré un effet inhibiteur de Twist sur la différenciation des gènes spécifiques du muscle [20, 21]. Dans le tissu osseux, Twist est également exprimé spécifiquement dans les cellules les plus immatures et son expression décroît considérablement à mesure que la différenciation ostéoblastique progresse, rendant de peu d’utilité les prélèvements osseux obtenus au cours d’opérations chirurgicales des patients. Une alternative consiste néanmoins à produire, à partir de ces échantillons de suture, des lignées ostéoblastiques immortalisées à l’aide de l’antigène T de SV-40. C’est en utilisant cette approche, à partir des ostéoblastes d’un malade n’exprimant plus que 50%de Twist, que l’équipe de P.J. Marie a récemment pu montrer que l’haplo-insuffisance de Twist se traduit par une expression accrue des marqueurs précoces du phénotype ostéoblastique (phosphatase alcaline et collagène de type I), associée à une nette augmentation de la formation osseuse [10]. Twist pourrait donc s’avérer un important modulateur négatif non seulement de la myogenèse mais également de l’ostéogenèse membranaire. Dans la mesure où les tissus musculaire et osseux possèdent la même origine mésodermique, il semble raisonnable d’imaginer que Twist puisse exercer un rôle dans le maintien de l’état indifférencié des cellules mésenchymateuses précurseurs, peut-être même avant que celles-ci ne soient engagées dans une voie de différenciation ostéoblastique. Cette hypothèse, que personne n’a encore vérifiée aujourd’hui, concorde avec deux observations récentes:la première indique que la surexpression de Twist dans des cellules ostéoblastiques de la lignée transformée HSaOs2 est capable d’induire la dé-différenciation de ces cellules vers un état de pré-ostéoblastes voire de précurseurs mésenchymateux n’exprimant plus aucun marqueur ostéoblastique [22]. Bien qu’il convienne de rester prudent quant à l’interprétation de phénotypes obtenus à partir de lignées transformées, Twist semble clairement influer sur l’expression des marqueurs ostéoblastiques précoces. La seconde observation rapporte une surexpression de Twist dans plusieurs rhabdomyosarcomes humains, tumeurs pédiatriques malignes qui affectent les cellules précurseurs du muscle squelettique qui ne sont pas entrées en différenciation, c’est-à-dire qui sont restées à l’état de précurseurs sans avoir été éliminées [23]. Une expression élevée de Twist semble donc bien associée à un phénotype mésenchymateux faiblement différencié et le maintien de cette expression pourrait être un obstacle majeur à toute différenciation ultérieure. Cependant, l’influence de Twist sur le maintien du statut mésenchymateux et les mécanismes moléculaires impliqués dans cette inhibition de la différenciation restent à élucider.
Conclusions et directions futures
Twist est le premier gène (et encore le seul à ce jour) dont une haploinsuffisance soit responsable d’une craniosténose syndromique. Avant lui, le facteur transcriptionnel à homéodomaine Msx2 et les récepteurs des FGF ont certes été associés à des craniosténoses syndromiques mais celles-ci étaient dues à des mutations hétérozygotes conférant un gain de fonction à la protéine (Tableau I) (m/s 1994, n°2, p. 225 et 1995, n° 12, p. 1748). A l’inverse, des délétions hétérozygotes du gène Msx2 ou des mutations inactivatrices de son homéodomaine ont été très récemment identifiées chez des patients présentant des foramens pariétaux, c’est-à-dire des lacunes osseuses de la voûte crânienne [24]. L’invalidation à l’état homozygote du gène Msx2 induit chez la souris un phénotype semblable, apparemment dû à un défaut de prolifération des cellules ostéoprogénitrices au niveau des fronts ostéogéniques [25] (m/s 2000, n°6-7, p. 816).
De même, on sait depuis peu qu’une haplo-insuffisance de Alx4, gène qui code pour un autre facteur transcriptionnel à homéoboîte exprimé dans le mésenchyme cranio-facial, est également responsable de foramens pariétaux [26, 27]. Ce phénotype «inverse de la craniosténose» est à rapprocher de la dysplasie cléido-crânienne, grave défaut d’ossification du crâne qui résulte de délétions ou de mutations ponctuelles hétérozygotes du gène Cbfa1 [28]. L’implication de ces différents facteurs dans des anomalies du développement crânien permet peu à peu de disposer, sur la scène de l’ostéoblaste, les différents acteurs impliqués dans sa différenciation précoce. Dès lors, la question de la fonction in vivo de Twist dans l’ossification de membrane se pose à la fois au plan moléculaire et au plan physiologique. Quelles sont les relations fonctionnelles entre ces différents protagonistes et comment Twist se situe-t-il dans la hiérarchie des facteurs qui influencent la différenciation du mésenchyme cranio-facial? Quelles sont les conséquences cellulaires d’une haplo-insuffisance de Twist susceptibles d’expliquer une fusion pathologique de la suture? Autant de questions qui restent en suspens et qui motivent actuellement l’analyse active des modèles cellulaires et animaux disponibles. L’idée commence néanmoins à poindre qu’une augmentation de l’apoptose locale au niveau de la suture puisse être impliquée dans sa synostose prématurée. D’intéressants résultats obtenus dans l’équipe de P.J. Marie suggèrent ainsi que l’haplo-insuffisance de Twist favorise l’apoptose dans les cellules ostéoblastiques humaines issues de calotte crânienne [29]. Dans ces cellules (immortalisées par l’antigène T de SV40), une surexpression du facteur pro-apoptotique Bax ainsi qu’une augmentation de l’activité des caspases 2, 8 et 3 ont été observées. Inversement, une surexpression de Twist est capable d’inhiber l’apoptose, induite par différents stimulus dans des fibroblastes d’embryons de souris [23], démontrant, dans ce contexte, l’effet anti-apoptotique de Twist. Bien qu’il reste à déterminer, parmi les cellules qui composent la suture, celles qui sont réellement le siège d’une mort cellulaire programmée, ce processus joue probablement un rôle important dans le devenir de la suture, c’est-à-dire son maintien ou sa fusion. La compréhension du rôle de Twist dans la dynamique de fusion de la voûte du crâne dépend à présent essentiellement du lien de cause à effet entre apoptose et synostose, et de l’identification de cibles directes in vivo de cet intéressant facteur de transcription
Références
- El Ghouzzi V, Le Merrer M, Perrin-Schmitt F, et al. Mutations of the TWIST gene in the Saethre-Chotzen syndrome. Nat Genet 1997; 15 : 42–6. [Google Scholar]
- Howard TD, Paznekas WA, Green ED, et al. Mutations in TWIST, a basic helix-loop-helix transcription factor, in Saethre-Chotzen syndrome. Nat Genet 1997; 15 : 36–41. [Google Scholar]
- Massari M, Murre C. Helix-loop-helix proteins : regulators of transcription in eucaryotic organisms. Mol Cell Biol 2000; 20 : 429–40. [Google Scholar]
- Bourgeois P, Bolcato-Bellemin AL, Danse JM, et al. The variable expressivity and incomplete penetrance of the twist-null heterozygous mouse phenotype resemble those of human Saethre-Chotzen syndrome. Hum Mol Genet 1998; 7 : 945–57. [Google Scholar]
- El Ghouzzi V, Lajeunie E, Le Merrer M, et al. Mutations within or upstream of the basic helix-loop-helix domain of the TWIST gene are specific to Saethre-Chotzen syndrome. Eur J Hum Genet 1999; 7 : 27–33. [Google Scholar]
- Gripp KW, Zackai EH, Stolle CA. Mutations in the human TWIST gene. Hum Mutat 2000; 15 : 150–5. [Google Scholar]
- Johnson D, Horsley SW, Moloney DM, et al. A comprehensive screen for TWIST mutations in patients with craniosynostosis identifies a new microdeletion syndrome of chromosome band 7p21.1. Am J Hum Genet 1998; 63 : 1282–93. [Google Scholar]
- El Ghouzzi V, Legeai-Mallet L, Aresta S, et al. Saethre-Chotzen mutations cause TWIST protein degradation or impaired nuclear location. Hum Mol Genet 2000; 9 : 813–9. [Google Scholar]
- El Ghouzzi V, Legeai-Mallet L, Benoist C, et al. Mutations in the basic domain and the loop-helix II junction of TWIST abolish DNA binding in Saethre-Chotzen syndrome. FEBS Lett 2001; 492 : 112–8. [Google Scholar]
- Yousfi M, Lasmoles F, Lomri A, Delannoy P, Marie PJ. Increased bone formation and decreased osteocalcin expression induced by reduced Twist dosage in Saethre-Chotzen syndrome. J Clin Invest 2001; 107 : 1153–61. [Google Scholar]
- Maquat LE, Carmichael GG. Quality control of mRNA function. Cell. 2001; 104 : 173–6. [Google Scholar]
- Johnson D, Iseki S, Wilkie AO, Morriss-Kay GM. Expression patterns of Twist and Fgfr1, -2 and -3 in the developing mouse coronal suture suggest a key role for twist in suture initiation and biogenesis. Mech Dev 2000; 91 : 341–5. [Google Scholar]
- Iseki S, Wilkie AO, Morriss-Kay GM. Fgfr1 and Fgfr2 have distinct differentiation-and proliferation-related roles in the developing mouse skull vault. Development 1999; 126 : 5611–20. [Google Scholar]
- Rice DP, Aberg T, Chan Y, et al. Integration of FGF and TWIST in calvarial bone and suture development. Development 2000; 127 : 1845–55. [Google Scholar]
- Delezoide AL, Benoist-Lasselin C, Legeai-Mallet L, et al. Spatio-temporal expression of FGFR 1, 2 and 3 genes during human embryo-fetal ossification. Mech Dev 1998; 77 77 : 19–30. [Google Scholar]
- Molteni A, Modrowski D, Hott M, Marie PJ. Differential expression of fibroblast growth factor receptor-1, -2, and -3 and syndecan-1, -2, and -4 in neonatal rat mandibular condyle and calvaria during osteogenic differentiation in vitro. Bone 1999; 24 : 337–47. [Google Scholar]
- Shishido E, Higashijima S, Emori Y, Saigo K. Two FGF-receptor homologues of Drosophila : one is expressed in mesodermal primordium in early embryos. Development 1993; 117 : 751–61. [Google Scholar]
- El Ghouzzi V, Quillet R, Stoetzel C, Munnich A, Bonaventure J, Perrin-Schmitt F. Analysis of the murine model of the Saethre-Chotzen syndrome suggests a functional relationship between TWIST and FGFR2. Bone 2001; 28 (suppl) : SC11W. [Google Scholar]
- Kophengnavong T, Michnowicz JE, Blackwell TK. Establishment of distinct MyoD, E2A, and twist DNA binding specificities by different basic region-DNA conformations. Mol Cell Biol 2000; 20 : 261–72. [Google Scholar]
- Hebrok M, Wertz K, Füchtbauer EM Mtwist is an inhibitor of muscle differentiation. Dev Biol 1994; 165 : 537–44. [Google Scholar]
- Hamamori Y, Wu HY, Sartorelli V, Kedes L. The basic domain of myogenic basic helix-loop-helix (bHLH) proteins is the novel target for direct inhibition by another bHLH protein, Twist. Mol Cell Biol 1997; 17 : 6563–73. [Google Scholar]
- Lee MS, Lowe GN, Strong DD, Wergedal JE, Glackin CA. TWIST, a basic helix-loop-helix transcription factor can regulate the human osteogenic lineage. J Cell Biochem. 1999; 75 : 566–77. [Google Scholar]
- Maestro R, Dei Tos AP, Hamamori Y, et al. Twist is a potential oncogene that inhibits apoptosis. Genes Dev 1999; 13 : 2207–17. [Google Scholar]
- Wilkie AO, Tang Z, Elanko N, et al. Functional haploinsufficiency of the human homeobox gene MSX2 causes defects in skull ossification. Nat Genet 2000; 24 : 387–90. [Google Scholar]
- Satokata I, Ma L, Ohshima H, et al. Msx2 deficiency in mice causes pleiotropic defects in bone growth and ectodermal organ formation. Nat Genet 2000; 24 : 391–5. [Google Scholar]
- Wuyts W, Cleiren E, Homfray T, Rasore-Quartino A, Vanhoenacker F, Van Hul W. The ALX4 homeobox gene is mutated in patients with ossification defects of the skull (foramina parietalia permagna, OMIM 168500). J Med Genet 2000; 37 : 916–20. [Google Scholar]
- Mavrogiannis LA, Antonopoulou I, Baxova A, et al. Haploinsufficiency of the human homeobox gene ALX4 causes skull ossification defects. Nat Genet. 2001; 27 : 17–8. [Google Scholar]
- Mundlos S, Otto F, Mundlos C, et al. Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell 1997; 89 : 773–9. [Google Scholar]
- Yousfi M, Marie PJ, Lasmoles F. Deletion of the basic helix-loop-helix domain in TWIST increases apoptosis in osteoblasts in the Saethre-Chotzen craniosynostosis. ASBMR, 22nd annual meeting, 2000. Toronto, Canada : ASBMR Abstract book, 2000. [Google Scholar]
- Sadler TW. Langman’s medical embryology, 7th ed. Baltimore (USA) : Lippincott-William and Wilkins, 1995. [Google Scholar]
Liste des tableaux
Les principales craniosténoses syndromiques transmises sur le mode autosomique dominant : dentification des gènes et des mécanismes impliqués.
Liste des figures
|
Figure 1. Représentation schématique du crâne d’un nouveau-né. Les différents os qui composent la voûte crânienne se rencontrent à la naissance sans pour autant fusionner complètement: les frontières entre deux os délimitent une suture alors que les carrefours entre plus de deux os forment une fontanelle (adapté d’après [30]). |
| Dans le texte | |
|
Figure 2. Le syndrome de Saethre-Chotzen résulte d’une haplo-insuffisance de Twist. A. Position des différentes mutations. Une cinquantaine de mutations du gène Twist (substitutions, mutations stop, délétions, insertions) ont été identifiées à l’etat hétérozygote chez de nombreux patients. Les mutations mentionnées en rouge prédisent la synthèse d’une protéine tronquée. B. Les différents niveaux d’haplo-insuffisance. L ’haploinsuffisance de Twist peut résulter de mécanismes distincts qui entrent en jeu à des étapes différentes de la synthèse ou de la fonction du facteur de transcription. Dans tous les cas, la mutation conduit finalement à une perte de fonction de Twist. |
| Dans le texte | |
|
Figure 3. Expression des gènes FGFR1, FGFR2, FGFR3 et Twist au niveau de la suture coronale chez la souris à 16 jours de développement. Twist est exprimé dans le tissu mésenchymateux qui constitue la suture, entre les fronts ostéogéniques. Ces derniers sont le siège d’une prolifération intense des pré-ostéoblastes qui est corrélee à une forte expression de FGFR2. Cette dernière, plus élevée encore dans le contexte d’une haplo-insuffisance de Twist, précède l’etape de différenciation osseuse au cours de laquelle FGFR1 devient plus exprimé que FGFR2 (Modifié d’après [13]). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.