")
")
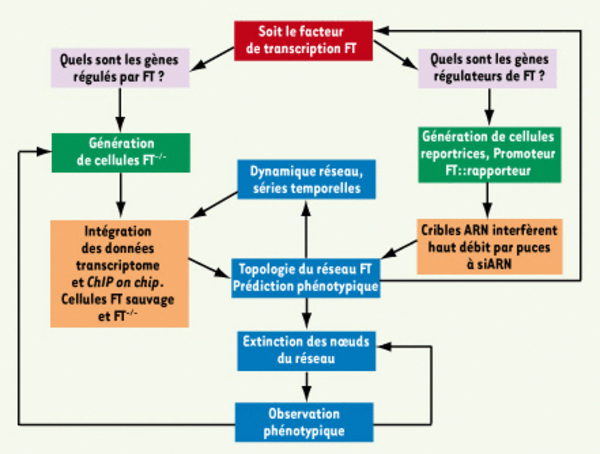
Figure 1.
Télécharger l'image originale
{kind=link}
Une approche systématique et intégrative pour inférer le réseau de régulation transcriptionnelle d’un facteur de transcription (FT) particulier. Afin d’identifier les gènes régulés par FT, des cellules délétées (cellules FT−/−) ou non (cellules sauvages) pour ce FT peuvent être utilisées pour analyser les modulations résultantes au niveau transcriptionnel (données transcriptome) et au niveau des cibles génomiques (données ChIP on chip). L’intégration de ces données génomiques peut alors permettre de construire la topologie du réseau en identifiant les gènes régulés par ce FT et d’étudier la dynamique de ce réseau, notamment via l’utilisation de séries temporelles. Pour identifier les régulateurs du FT, une stratégie différente est envisagée. Des cellules contenant une construction reportrice dans laquelle le promoteur du FT est fusionné à un gène rapporteur sont utilisées pour cribler à haut débit des collections de siARN sur puce. L’identification des gènes dont l’extinction entraîne une induction ou une répression de l’expression du FT permet de compléter la topologie du réseau, en identifiant les gènes régulant le FT. L’ensemble des informations collectées sur la topologie du réseau peut également servir de base pour identifier les partenaires clés du FT dans le réseau. Ces partenaires peuvent à leur tour être éliminés et servir à générer de nouvelles données génomiques qui viendront enrichir la topologie du réseau établie précédemment. L’impact du réseau sur le phénotype des cellules est étudié par extinction systématique des nœuds du réseau.
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.