")
")
| Issue |
Med Sci (Paris)
Volume 17, Number 11, Novembre 2001
|
|
|---|---|---|
| Page(s) | 1168 - 1175 | |
| Section | Hypothèses/Débats | |
| DOI | https://doi.org/10.1051/medsci/200117111168 | |
| Published online | 15 novembre 2001 | |
Évolution des séquences « signal de recombinaison » dans le locus de la région variable de la chaîne lourde des immunoglobulines
Evolution of the recombination signal sequences in the immunoglobulin heavy chain variable region locus
Service de biosystématique, Université Pierre-et-Marie-Curie (Paris 6), Bât. B, 7 e étage, case 241, 9, quai St-Bernard, 75252 Paris Cedex 05, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Les locus des immunoglobulines (Ig) et des récepteurs des cellules T (TcR) sont propres aux vertébrés et présentent une dynamique évolutive exceptionnelle dont les modalités restent cependant à découvrir. Les gènes des Ig et TcR sont assemblés au cours du développement des lymphocytes, à partir des segments codants V, D, et J, par un mécanisme spécifique appelé recombinaison V(D)J. Ce mécanisme très particulier fait intervenir de courtes séquences cibles adjacentes aux segments codants appelées séquences « signal de recombinaison » (SSR). Des travaux récents ont montré que la recombinaison V(D)J pouvait avoir lieu dans les cellules germinales des poissons cartilagineux [12]. Si un tel mécanisme était avéré chez les mammifères, il pourrait expliquer les importantes restructurations observées dans les locus des Ig et des TcR au cours de l’évolution. Dans le cadre de cette hypothèse, on s’attendrait alors à ce que les segments codants et les SSR n’aient pas toujours évolué comme une unité fonctionnelle. Autrement dit, des analyses phylogénétiques menées indépendamment surles segments codants et leurs SSR adjacentes devraient montrer des histoires évolutives différentes.
L’étude du locus de la région variable de la chaîne lourde des Ig a permis de définir quatre types de SSR chez l’homme : H1, H2, H3 et H5. L’analyse des séquences disponibles dans les banques de données a indiqué que les autres mammifères ne présentaient pas de types de SSR autres que ceux découverts chez l’homme. Les analyses phylogénétiques menées sur les segments codants VH et leurs SSR adjacentes ont montré qu’il y a eu remplacement d’une SSR de type H2 par une SSR de type H3 au cours de l’évolution des mammifères. Deux modèles sont proposés pour rendre compte de ce remplacement : le premier implique un crossing-over inégal, le second fait intervenir une activation de la recombinaison V(D)J dans les cellules germinales. Ce remplacement semble avoir été sélectionné positivement au cours de l’évolution des mammifères car les SSR H3 apparaissent plus efficaces pour la recombinaison V(D)J que les SSR H2.
Abstract
The immunoglobulin (Ig) and T cell receptor (TCR) loci of vertebrates present an exceptionally dynamic evolutionary history, but the mechanisms responsible remain unknown. Genes encoding the V, D, and J segments of antigen receptors are somatically rearranged in developing lymphocytes by a specialised DNA recombination mechanism called V (D) J recombination. This sitespecific recombination requires the presence of short target sequences, named « Recombination Signal Sequences » (RSS), which are adjacent to the coding segments V, D and J. Recently, V (D) J recombination has been reported in the germline of cartilaginous fishes. Assuming that germline V (D)J rearrangements also occurred during the mammal history, we expect to find that coding segments and RSSs were not always associated as a functional unit during evolution. In other words, the phylogenetic tree constructed from coding segments could be different from the one produced with the RSSs. This possibility is here examined by studying the complete human Ig heavy chain variable region (VH) locus. In this locus, four distinct types of RSS have been defined, namely H1, H2, H3, and H5. No other RSS type has been detected in other mammalian species. The phylogenetic analyses, performed on one hand on the coding VH segments and on the other hand on the adjacent RSSs, revealed that the type H2 RSS of one VH gene has clearly become replaced by a type H3 RSS during mammalian evolution. Two general models are proposed to explain the RSS swap : the first involves an unequal crossingover, the second implicates germline activation of V (D)J recombination. The recombination event has likely been selected during the evolution of mammals because the type H3 RSS seem to provide better VDJ recombination efficiency than the type H2 RSS.
© 2001 médecine/sciences - Inserm / SRMS
Le système immunitaire des vertébrés produit un vaste répertoire d’immunoglobulines (Ig) et de récepteurs des cellules T (TcR : T cell receptor) capables de reconnaître un très large spectre d’antigènes. Une molécule d’Ig est un tétramère polypeptidique composé de deux chaînes légères (L) identiques et de deux chaînes lourdes (H) identiques (figure 1). Une molécule de TcR est un hétérodimère composé soit d’une chaîne α et d’une chaîne β, soit d’une chaîne γ et d’une chaîne δ. Les chaînes polypeptidiques des Ig et des TcR sont constituées d’une région variable responsable des interactions avec l’antigène et d’une région constante. La région constante est codée par un seul segment de gène. La région variable est codée par une séquence d’ADN assemblée soit à partir de deux segments de gène (un segment V [variable] et un segment J [jonction]) dans le cas des chaînes L, α et γ, soit à partir de trois segments de gne (V, D [diversité] et J) dans le cas des chaînes H, β et δ. Dans le génome des vertébrs, il existe un grand nombre de segments V, D et J et, en théorie, chaque segment V peut être combiné avec n’importe quel segment D et n’importe quel segment J. Ce mécanisme combinatoire, appelé recombinaison VDJ, permet d’obtenir une très grande diversité de réarrangements. Ces réarrangements s’effectuent uniquement dans les lignées lymphocytaires B et T et permettent l’établissement des répertoires somatiques des Ig et TcR, propres à chaque individu [1, 2].
 |
Figure 1. La recombinaison VDJ dans le locus de la chaîne lourde des immu-noglobulines. Les séquences « signal de recombinaison » (SSR) sont représentées sous la forme d’un triangle rouge; celles situées en 3’ des segments V et D sont caractérisées par un espaceur de 23 pb alors que celles localisées en 5’ des segments D et J présentent un espaceur de 12 pb. Généralement, la recombinaison a lieu entre des SSR de tailles différentes et orientées en sens inverse (règle 12/23) [1, 2]. |
Les réarrangements entre les éléments V, D et J sont réalisés grâce à de courtes séquences cibles, appelées séquences « signal de recombinaison» (SSR), situées immédiatement en 3’ des segments V, en 5’ et 3’ des segments D, et en 5’ des segments J (figure 1). Les SSR sont composées de deux motifs très conservés (un heptamère et un nonamère) séparés par un «espaceur» de 12 ou 23 nucléotides. Les réarrangements VDJ sont provoqués par l’activité des protéines RAG-1 et RAG-2 (recombination activating gene), qui sont capables de reconnaître les SSR et d’engendrer des coupures d’ADN double brin entre les SSR et les segments de gène V, D et J [1, 2].
SSR associées aux segments de gène VH
Chez l’homme, tous les segments de gène VH de la chaîne lourde des immunoglobulines sont connus puisque le locus de la région variable a été complètement séquencé [3]. Une étude restreinte aux 39 segments VH fonctionnels du locus humain a permis de définir quatre types de SSR, appelés H1, H2, H3, et H5 [4]. Ces quatre types de SSR sont facilement différentiables en comparant soit les espaceurs de 23 pb des SSR, soit les régions non fonctionnelles situées en 3’ des SSR (figure 2). Il est possible d’aligner les régions 3’-adjacentes des SSR lorsque les séquences comparées appartiennent à un même type de SSR. En revanche, il s’avère impossible de les aligner lorsque les séquences comparées appartiennent à des types de SSR différents.
 |
Figure 2. Alignement des 39 SSR fonctionnelles du locus VH humain. L’alignement inclut les trois régions des SSR (heptamère, espaceur de 23 nucléotides et nonamère) ainsi que la séquence 3’-adjacente. Toutes ces données sont tirées de la séquence complète du locus VH humain [3]. Chez l’Homme, les segments de gène VH ont été classés en sept familles (la famille 7 n’est pas représentée ici car aucun segment fonctionnel n’y a été décrit) qui ont été rassemblées dans trois groupes majeurs (A en bleu, B en rouge et C en vert) [5-7]. Le premier chiffre de chaque nom de segment VH fait référence à sa famille d’appartenance, le second chiffre indique sa position sur le chromosome 14 [3]. Pour la nomenclature des SSR, la lettre H fait référence à l’Homme alors que le chiffre indique la famille VH la plus représentative de cet ensemble [4]. Le type H3 est le plus commun avec 26 membres correspondant aux familles VH 3, 4 et 6. Le type H2 est associé à la famille VH 2. Le type H1 inclut tous les représentants de la famille VH 1. Le type H5 ne contient que VH 5-51. |
Toutes les SSR associées aux segments VH de mammifères appartiennent aux types H1 et H3 mis en évidence dans le locus VH humain. Le type H1 a été retrouvé uniquement chez la souris, alors que le type H3 est beaucoup plus répandu : il a été détecté chez des cétartiodactyles (bœuf, mouton, chameau et porc), un lagomorphe (lapin), des primates (chimpanzé, gorille et macaque) et un rongeur (souris). Aucune SSR de mammifère ne ressemble aux SSR découvertes chez les oiseaux, les reptiles, les amphibiens et les poissons. Ce résultat s’explique probablement par la longue période de temps qui s’est écoulée depuis la séparation des principaux groupes de vertébrés, ce qui a entraîné l’accumulation d’un grand nombre de mutations dans les régions utilisées pour définir les différents types de SSR (l’espaceur et la région 3’-adjacente de la SSR), obscurcissant ainsi le signal phylogénétique ancien.
Le modèle d’évolution birth-and-death
Le modèle d’évolution birth-and-death postule que les nouveaux segments VH sont créés par duplication ; certains sont maintenus sous forme fonctionnelle dans le génome, d’autres sont éliminés ou deviennent des pseudogènes à la suite de mutations délétères [5, 6]. Pour donner naissance à un nouveau segment de gène fonctionnel, le fragment d’ADN dupliqué doit inclure la séquence VH codante, sa SSR 3’-adjacente, ainsi que la région 5’-régulatrice de la transcription. Ces trois éléments ont donc probablement évolué comme une unité fonctionnelle, ce qui implique qu’ils partagent la même histoire phylogénétique. Cette hypothèse a été testée en analysant la distribution des différents types de SSR sur l’arbre phylogénétique obtenu avec les séquences VH (figure 3).
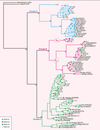 |
Figure 3. Arbre phylogénétique des segments de gène VH de mammifères. Les analyses phylogénétiques ont été réalisées avec le logiciel PAUP 3.1.1 [15] en examinant l’exon 2 (261 pb) de 85 segments VH de mammifères. L’arbre correspond au consensus strict des 54 arbres les plus courts (longueur = 396 410 pas) obtenus en parcimonie pondérée [16, 17]. La robustesse des regroupements a été estimée par la méthode du Bootstrap [18]: les valeurs obtenues après 100 réplications sont indiquées sur les branches. Les trois principaux groupes de séquences VH sont distingués par un code couleur : groupe A en bleu, groupe B en rouge et groupe C en vert. À l’extrémité des branches, les cercles indiquent que la SSR associée au segment VH est connue, la couleur du cercle précise le type de SSR concerné. Les séquences autres qu’humaines sont désignées par le nom de genre de l’animal suivi par le numéro d’accès dans Genbank. L’arbre a été enraciné avec les segments VH 101 de requin (Heterodontus francisci) et VH 102 de raie (Raja erinacea), car ces séquences se branchent avant la divergence des segments VH de mammifères [5]. |
Conformément à de précédentes analyses [5-7], trois groupes majeurs de séquences VH, A, B et C, ont été mis en évidence chez les mammifères (figure 3). La présence de séquences VH appartenant à la fois à l’homme et à la souris dans ces trois groupes suggère que leur séparation est antérieure à la radiation des mammifères euthériens, il y a 65 millions d’années (MA) [7]. D’autres analyses ont montré, par ailleurs, que les groupes A et B contiennent des amphibiens en plus des mammifères, alors que le groupe C inclut aussi des oiseaux, des reptiles, des amphibiens, et des poissons osseux [5, 6]. Ceci indique que les groupes A, B et C avaient déjà divergé avant la radiation des tétrapodes aux alentours de 350-400 MA [5].
Selon le modèle birth-and-death, les trois groupes A, B et C obtenus sur la base des séquences VH devraient être retrouvés en analysant les SSR. Ce modèle se vérifie dans le cas des groupes A et C (figure 3) : les segments VH du groupe A sont flanqués de SSR de types H1 ou H5, lesquels apparaissent apparentés sur la base des séquences de l’espaceur (mais pas sur la base des régions 3’-adjacentes des SSR, ce qui justifie la définition de deux types différents (figure 2), et tous les segments VH du groupe C sont liés à des SSR appartenant au seul type H3. En revanche, l’analyse au sein du groupe B ne permet pas d’avaliser le modèle birth-and-death car les segments VH sont associés à des SSR de deux types non apparentés, H2 et H3 (correspondant respectivement aux groupes BH2 et BH3, figure 3).
Quelle est l’origine des SSR H3 du groupe B ?
Deux hypothèses permettent d’interpréter la présence de SSR de type H3 dans le groupe B (figure 4). Selon la première (Hypothèse H3), le segment VH ancestral du groupe B était connecté à une SSR H3 et l’ancêtre du groupe BH2 s’est doté d’une SSR du type H2 par un processus qui reste à déterminer. Inversement, la seconde (Hypothèse H2) stipule que le segment VH ancestral du groupe B était lié à une SSR H2 et que l’ancêtre du groupe BH3 a obtenu une SSR du type H3 à partir d’un donneur VH du groupe C.
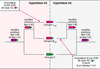 |
Figure 4. Deux hypothèses pour expliquer l’origine des SSR H3 du groupe B. La première (Hypothèse H3), suppose que le segment VH ancestral du groupe B était connecté à une SSR H3 et que l’ancêtre du groupe BH2 s’est doté d’une SSR du type H2 par un processus qui reste à déterminer. Ceci implique a fortiori que le type de SSR H3 était déjà différencié chez l’ancêtre commun de toutes les séquences VH des groupes B et C. Inversement, la seconde (Hypothèse H2) stipule que le segment VH ancestral du groupe B é tait lié à une SSR H2 et que l’ancêtre du groupe BH3 a obtenu une SSR du type H3 à partir d’un donneur VH du groupe C. Les segments de gène VH sont représentés par des rectangles rouges pour ceux du groupe B et verts pour ceux du groupe C. Les SSR sont symbolisées par des triangles rouges pour celles du type H2, et verts pour celles du type H3. La couleur violette est utilisée pour représenter les séquences ancestrales communes aux VH des groupes B et C. |
Trois arguments soutiennent cette dernière hypothèse et indiquent que l’histoire évolutive du locus VH a été marquée par un échange de SSR entre deux segments VH des groupes B et C. (1) Puisque les groupes B et C avaient déjà divergé avant la radiation des tétrapodes [5], l’hypothèse H3 suppose une conservation tout à fait exceptionnelle des SSR et des régions 3’-adjacentes. Si tel était le cas, on s’attendrait logiquement à trouver non seulement les SSR de type H3 dans tous les segments VH des groupes B et C de mammifères, mais aussi dans ceux d’autres groupes de tétrapodes. Pourtant, aucun des segments des groupes B et C décrits à ce jour chez les oiseaux, les crocodiles et les amphibiens ne présente de SSR H3. (2) Sous l’hypothèse H3, il faut supposer que le type H2 provient d’un donneur d’origine inconnue. Dans le cas de l’hypothèse H2, l’origine du type H2 est connue : il s’agit du type ancestral du groupe B, lequel est le résultat d’un simple processus de divergence nucléotidique à partir de la SSR ancestrale des groupes B et C. (3) La comparaison des groupes BH3 et C révèle que les SSR et les régions 3’-adjacentes sont très proches alors que les régions situées en 5’ des SSR ne sont pas apparentées (région 5’ régulatrice, exon 1, intron et exon 2 des segments VH). De telles différences ne s’expliquent pas par un simple processus de mutation nucléotidique. Seul un événement de recombinaison peut rendre compte de ces différences.
Bien qu’il soit difficile de dater précisément l’événement qui a conduit au remplacement d’une SSR H2 par une SSR H3, trois indices reposant sur des données paléontologiques [8] indiquent qu’il a pu avoir lieu entre 115 et 65 MA. (1) L’absence de SSR H3 chez les amphibiens, reptiles et oiseaux, suggère que l’événement est arrivé après l’émergence des premiers mammifères, aux alentours de 200 MA. (2) Le groupe BH3 étant composé d’espèces appartenant à trois ordres différents de mammifères (cétartiodactyles, primates et rongeurs), cela indique que l’événement a pris place avant la radiation adaptative des mammifères euthériens, il y a 65 MA. (3) L’analyse des ADN complémentaires de marsupiaux disponibles dans les banques de données montre qu’aucune séquence VH de Monodelphis domestica et Trichosurus vulpecula n’est liée au groupe B, ce qui laisse penser que l’événement a eu lieu après l’émergence des mammifères euthériens, il y a environ 115 MA.
Les SSR de type H3 sont-elles plus efficaces pour la recombinaison VDJ ?
Chez l’Homme, la grande majorité des segments VH fonctionnels sont associés à des SSR H3 (26 sur un total de 39 séquences, figure 2) alors que les SSR H2 sont très peu représentées (seulement trois exemplaires). Par ailleurs, il est frappant de constater qu’à ce jour toutes les espèces de mammifères pour lesquelles des SSR ont été séquencées possèdent le type H3 (chameau, lapin, mouton, porc et souris), alors que le type H2 n’a été décrit que chez l’homme. De plus, des analyses phylogénétiques incluant les séquences d’ADN complémentaire ont révélé d’autres espèces susceptibles de détenir des SSR H3 (cheval, chien, lama, opossums d’Amérique du Sud et d’Australie, rat et vison). Ces mêmes analyses laissent toutefois penser que des SSR H2 pourraient être découvertes dans le génome du rat et de la souris.
Le succès évolutif des SSR H3 contraste fortement avec la rareté des SSR H2 au sein des mammifères. La comparaison des SSR humaines indique que cette représentation limitée pourrait s’expliquer par l’atypisme de l’heptamère et du nonamère des SSR H2. Alors que l’heptamère CACAGTG caractérise la grande majorité des SSR (toutes celles des types H1 et H5, ainsi que 24 des 26 SSR du type H3), il est surprenant de constater que l’heptamère du type H2 correspond à CACAAAG ou CACAGAG (figure 2). Des tests fonctionnels ont en effet montré que l’altération de n’importe lequel des nucléotides de l’heptamère CACAGTG entraîne une forte diminution dans les fréquences de recombinaison [9, 10]. Concernant le nonamère, le motif AAA en position 5-7 apparaît primordial pour assurer la fonction de recombinaison [9,10]. Ce motif est conservé chez tous les SSR H1, H3 et H5, et une fois encore le type H2 apparaît exceptionnel, deux de ses trois représentants ne possédant pas ce motif (figure 2). Les variations de l’heptamère et du nonamère observées dans les SSR H2 suggèrent donc une faible efficacité de ces séquences pour la recombinaison VDJ. En accord avec cette hypothèse, l’analyse des IgM exprimées dans les cellules B humaines circulantes [11] a montré qu’au sein du groupe B, les séquences VH associées à des SSR H2 étaient sous-exprimées par rapport à leur représentation dans le génome (expression attendue 7,7 %, expression observée < 3 %), alors que les séquences VH liées à des SSR H3 étaient davantage exprimées (famille VH4 : attendue 15,4 %, observée 19-23 %).
En conclusion, le remplacement d’une SSR H2 par une SSR H3 au cours de l’évolution des mammifères a probablement été sélectionné positivement car les SSR H3 apparaissent plus efficaces pour la recombinaison VDJ. En faveur de cette hypothèse, il faut aussi noter que toutes les espèces caractérisées par un répertoire restreint de segments VH (chameau, porc, lapin et mouton) ne présentent que des SSR de type H3 (figure 3).
Comment s’est produit l’échange de SSR au cours de l’évolution des mammifères ?
Deux modèles peuvent expliquer la présence de deux types de SSR (H2 et H3) au sein du groupe B (figure 5). Le premier modèle fait intervenir un crossing-over inégal dans les cellules germinales qui aurait entraîné une duplication en tandem sur le chromosome sélectionné au cours de l’évolution (figure 5, I). Ce modèle suppose que la recombinaison homologue puisse se faire entre des séquences VH divergentes, les groupes B et C étant séparés depuis plus de 350-400 MA [5].
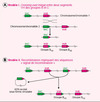 |
Figure 5. Deux modèles pour expliquer la présence de SSR H3 dans le groupe B. Le premier fait intervenir un crossing-over inégal (modèle I), le second implique un événement de type recombinaison V (D)J (modèle II). Les segments de gène VH sont représentés par des rectangles rouges pour ceux du groupe B et verts pour ceux du groupe C. Les SSR sont symbolisées par des triangles rouges pour celles du type H2, et verts pour celles du type H3. |
Le second modèle suppose que l’échange de SSR s’est produit par un mécanisme faisant intervenir une reconnaissance des SSR par les protéines RAG (figure 5, II). Ce modèle suppose donc que la recombinaison de type VDJ puisse exceptionnellement avoir lieu dans les cellules germinales des mammifères, comme cela a été montré chez les requins [12, 13]. Cependant, au lieu d’obtenir une recombinaison standard, c’est-à-dire impliquant deux SSR de tailles différentes (l’une avec un espaceur de 23pb et l’autre avec un espaceur de 12pb) et orientées en sens inverse, le modèle proposé (figure 5, II) prédit que la recombinaison a été réalisée entre deux SSR présentant une taille et une orientation identique. Cette situation particulière ayant déjà été observée expérimentalement [14], rien ne permet d’exclure cette éventualité, d’autant que le site apparent de recombinaison est localisé au niveau de la limite entre la séquence VH et la SSR.
Indépendamment du mécanisme impliqué, il est clair que la possibilité de changement de SSR a été un élément important au cours de l’évolution du locus VH des mammifères. Par ce moyen, des segments de gène présentant de meilleures propriétés de liaison à l’antigène pourraient en effet acquérir un niveau de représentation plus élevé dans le répertoire du système immunitaire

Remerciements
Je remercie le Pr Jean Deutsch et les Drs Rachel Golub et Michaël Manuel pour leurs commentaires sur la première version du manuscrit.
Références
- Lewis SM. The mechanism of V(D)J joining : lessons from molecular, immunological, and comparative analyses. Adv Immunol 1994; 56 : 27–150. [Google Scholar]
- Gellert M. Recent advances in understanding V(D)J recombination. Adv Immunol 1997; 64 : 39–64. [Google Scholar]
- Matsuda F, Ishii K, Bourvagnet P, et al. The complete nucleotide sequence of the human immunoglobulin heavy chain variable region locus. J Exp Med 1998; 188 : 2151–62. [Google Scholar]
- Hassanin A, Golub R, Lewis SM, Wu GE. Evolution of the recombination signal sequences in the Ig heavy-chain variable region locus of mammals. Proc Natl Acad Sci USA 2000; 97 : 11415–20. [Google Scholar]
- Ota T, Nei M. Divergent evolution and evolution by the birth-and-death process in the immunoglobulin VH gene family. Mol Biol Evol 1994; 11 : 469–82. [Google Scholar]
- Nei M, Gu X, Sitnikova T. Evolution by the birth-and-death process in multigene families of the vertebrate immune system. Proc Natl Acad Sci USA 1997; 94 : 7799–806. [Google Scholar]
- Schroeder HWJ, Hillson JL, Perlmutter RM. Structure and evolution of mammalian VH families. Int Immunol 1990; 2 : 41–50. [Google Scholar]
- Benton MJ. Vertebrate Palaeontology. Second Édition, Chapman & Hall, London, 1997 : 452 p. [Google Scholar]
- Hesse JE, Lieber MR, Mizuuchi K, Gellert M. V(D)J recombination : a functional definition of the joining signals. Genes Dev 1989; 3 : 1053–61. [Google Scholar]
- Akamatsu Y, Tsurushita N, Nagawa F, et al. Essential residues in V(D)J recombination signals. J Immunol 1994; 153 : 4520–9. [Google Scholar]
- Rassenti LZ, Kohsaka H, Kipps TJ. Analysis of immunoglobulin VH gene repertoire by an anchored PCR-ELISA. Ann NY Acad Sci 1995; 764 : 463–73. [Google Scholar]
- Lee SS, Fitch D, Flajnik MF, Hsu E. Rearrangement of immunoglobulin genes in shark germ cells. J Exp Med 2000; 191 : 1637–48. [Google Scholar]
- Litman GW, Anderson MK, Rast JP. Evolution of antigen binding receptors. Annu Rev Immunol 1999; 17 : 109–47. [Google Scholar]
- Lewis SM, Hesse JE, Mizuuchi K, Gellert M. Novel strand exchanges in V(D)J recombination. Cell 1988; 55 : 1099–107. [Google Scholar]
- Swofford DL. PAUP : Phylogenetic Analysis Using Parsimony, version 3.1.1, Illinois Natural History Survey, Champaign IL, 1993. [Google Scholar]
- Hassanin A, Lecointre G, Tillier S. The « evolutionary signal » of homoplasy in protein-coding gene sequences and its consequences for a priori weighting in phylogeny. CR Acad Sci III 1998; 321 : 611–20. [Google Scholar]
- Hassanin A, Pasquet E, Vigne JD. Systematic relationships within the subfamily Caprinae (Artiodactyla, Bovidae) as determined from cytochrome b sequences. J Mammal Evol 1998; 5 : 217–36. [Google Scholar]
- Felsenstein J. Confidence limits on phylogenies : an approach using the bootstrap. Evolution 1985; 39 : 783–91. [Google Scholar]
Liste des figures
|
Figure 1. La recombinaison VDJ dans le locus de la chaîne lourde des immu-noglobulines. Les séquences « signal de recombinaison » (SSR) sont représentées sous la forme d’un triangle rouge; celles situées en 3’ des segments V et D sont caractérisées par un espaceur de 23 pb alors que celles localisées en 5’ des segments D et J présentent un espaceur de 12 pb. Généralement, la recombinaison a lieu entre des SSR de tailles différentes et orientées en sens inverse (règle 12/23) [1, 2]. |
| Dans le texte | |
|
Figure 2. Alignement des 39 SSR fonctionnelles du locus VH humain. L’alignement inclut les trois régions des SSR (heptamère, espaceur de 23 nucléotides et nonamère) ainsi que la séquence 3’-adjacente. Toutes ces données sont tirées de la séquence complète du locus VH humain [3]. Chez l’Homme, les segments de gène VH ont été classés en sept familles (la famille 7 n’est pas représentée ici car aucun segment fonctionnel n’y a été décrit) qui ont été rassemblées dans trois groupes majeurs (A en bleu, B en rouge et C en vert) [5-7]. Le premier chiffre de chaque nom de segment VH fait référence à sa famille d’appartenance, le second chiffre indique sa position sur le chromosome 14 [3]. Pour la nomenclature des SSR, la lettre H fait référence à l’Homme alors que le chiffre indique la famille VH la plus représentative de cet ensemble [4]. Le type H3 est le plus commun avec 26 membres correspondant aux familles VH 3, 4 et 6. Le type H2 est associé à la famille VH 2. Le type H1 inclut tous les représentants de la famille VH 1. Le type H5 ne contient que VH 5-51. |
| Dans le texte | |
|
Figure 3. Arbre phylogénétique des segments de gène VH de mammifères. Les analyses phylogénétiques ont été réalisées avec le logiciel PAUP 3.1.1 [15] en examinant l’exon 2 (261 pb) de 85 segments VH de mammifères. L’arbre correspond au consensus strict des 54 arbres les plus courts (longueur = 396 410 pas) obtenus en parcimonie pondérée [16, 17]. La robustesse des regroupements a été estimée par la méthode du Bootstrap [18]: les valeurs obtenues après 100 réplications sont indiquées sur les branches. Les trois principaux groupes de séquences VH sont distingués par un code couleur : groupe A en bleu, groupe B en rouge et groupe C en vert. À l’extrémité des branches, les cercles indiquent que la SSR associée au segment VH est connue, la couleur du cercle précise le type de SSR concerné. Les séquences autres qu’humaines sont désignées par le nom de genre de l’animal suivi par le numéro d’accès dans Genbank. L’arbre a été enraciné avec les segments VH 101 de requin (Heterodontus francisci) et VH 102 de raie (Raja erinacea), car ces séquences se branchent avant la divergence des segments VH de mammifères [5]. |
| Dans le texte | |
|
Figure 4. Deux hypothèses pour expliquer l’origine des SSR H3 du groupe B. La première (Hypothèse H3), suppose que le segment VH ancestral du groupe B était connecté à une SSR H3 et que l’ancêtre du groupe BH2 s’est doté d’une SSR du type H2 par un processus qui reste à déterminer. Ceci implique a fortiori que le type de SSR H3 était déjà différencié chez l’ancêtre commun de toutes les séquences VH des groupes B et C. Inversement, la seconde (Hypothèse H2) stipule que le segment VH ancestral du groupe B é tait lié à une SSR H2 et que l’ancêtre du groupe BH3 a obtenu une SSR du type H3 à partir d’un donneur VH du groupe C. Les segments de gène VH sont représentés par des rectangles rouges pour ceux du groupe B et verts pour ceux du groupe C. Les SSR sont symbolisées par des triangles rouges pour celles du type H2, et verts pour celles du type H3. La couleur violette est utilisée pour représenter les séquences ancestrales communes aux VH des groupes B et C. |
| Dans le texte | |
|
Figure 5. Deux modèles pour expliquer la présence de SSR H3 dans le groupe B. Le premier fait intervenir un crossing-over inégal (modèle I), le second implique un événement de type recombinaison V (D)J (modèle II). Les segments de gène VH sont représentés par des rectangles rouges pour ceux du groupe B et verts pour ceux du groupe C. Les SSR sont symbolisées par des triangles rouges pour celles du type H2, et verts pour celles du type H3. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.